


ABSTRACT
Background: The function of the intervertebral disc is structural. Loss of tissue alters biomechanics, leads to subsequent disc degeneration, and is attributable to discogenic pain. A viable structural allograft was delivered into degenerate discs to determine whether intervention could safely stabilize anatomy, reduce pain, and improve function.
Methods: Following institutional review board approval and patient consent, subjects were randomized to receive allograft or saline at either 1 or 2 levels or continue nonsurgical management (NSM). Data were collected at baseline, 3, 6, and 12 months. Back pain with a visual analog scale (VAS) and disability by the Oswestry Disability Index (ODI) were assessed, as were adverse events. This trial is registered on http://www.clinicaltrials.gov (NCT03709901).
Results: At 6 and 12 months, the VAS improved from 54.81, 55.25, and 62.255 in the allograft, saline, and NSM subjects, respectively, to 16.0 and 41.0 in the allograft and saline groups at 6 months, and 12.27 and 19.67, respectively, at 12 months. All subjects in the NSM cohort crossed over to allograft treatment. At 6 and 12 months, ODI improved from 53.73, 49.25, and 55.75 in the allograft, saline, and NSM subjects, respectively, to 18.47 and 28.75 in the allograft and saline groups 1 and 2 at 6 months, and 15.67 and 9.33, respectively, at 12 months. At 3 months the ODI of the NSM group was 62.75 and subjects reached 19.0 and 11.0 at 6 and 12 months, respectively. Adverse events were transient and resolved in all cohorts.
Conclusions: This study is supported by data demonstrating that improved pain and function at 12 months can be attained with a supplemental viable disc matrix. Subjects receiving the VIA Disc Matrix achieved improvements that were durable at 12 months.
Level of Evidence: 1.
Clinical Relevance: Initial assessments indicate that a 1-level or 2-level treatment offers a reliable intervention that is safe and beneficial.
INTRODUCTION
Vertebrates arose more than 530 million years ago, and the evolution of vertebral bone from the flexible notochord was a key event that provided foundation morphology supporting a structural anatomy that renders the spine stiffer while bending. This attribute of bending without shortening contributes to important mechanical features that enable storing and releasing energy elastically like a spring.
The function of the intervertebral disc is predominantly structural, supporting upright posture without compromising freedom of rotation, flexion, or compression. With such degrees of freedom, it is the perfect tissue to support bipedal anatomy and the active use of upper extremities while standing or ambulating. The advent of flexibility comes with a parallel morphology that is unique to the intervertebral disc, which contains structured water that carries hydraulic qualities, and mechanically plastic properties that simultaneously support movement and resistance to stress. Retaining the notochordal qualities centrally as a hallmark of elasticity in the nucleus pulposus, the evolutionary stiffening of the vertebral bodies created a flexible load bearing structure that has unfortunately made the human spine particularly prone to pathology.
A high concentration of proteoglycans in the nucleus pulposus produces an intradiscal pressure that is imperative to the mechanical function of the disc. Unfortunately for sustaining long-term function, the disc has a low capacity for replenishment. It is alymphatic, aneural, and avascular; therefore, it is dependent on transfer of nutrients to and metabolic waste across the subchondral plate. Over time, degeneration takes its toll on the structure, with a loss of proteoglycans leading to a loss of intradiscal pressure due to the decreasing ability to hold structured water and the loss of functional tissue and altered mechanics. Over time the tissue loss and resultant mechanical alteration of the disc renders the intervertebral disc unstable. The inherent degrees of freedom that are assets to disc mechanics in healthy tissue are liabilities under degenerate conditions. Laxity of the spine can produce spondylolisthesis and reduce disc height; pain emerges as a result of this instability and abnormal force transmission.
Low back pain is the leading cause of disability in developed countries, with the number of people affected worldwide increasing annually.1 Degenerative disc disease is a major factor contributing to this disability and is the most common etiology of chronic lower back pain in adults. More than 40% of the adult population of the United States reported low back pain at some point within the preceding 3 months prior to them being questioned and this has a substantial socioeconomic impact on the affected population.2,3 Causal factors vary considerably from the innocuous insult of repeated heavy lifting or sudden awkward movement resulting in an acute injury to the more insidious condition of disc degeneration. Though several risk factors have been identified (including occupational posture, depressive moods, obesity, body height, and age), the genesis of low back pain remains obscure. Back pain is not a disease but a constellation of symptoms, and in most cases the origins remain unknown.
Low back pain affects people of all ages, from children to the elderly. Estimating the initial back pain incidence is challenging due to a relatively high prevalence of back pain in early adulthood that persists with recurrent symptoms over time. That noted, the lifetime prevalence of nonspecific (common) low back pain has been estimated at 60% to 70% in industrialized countries (1-year prevalence, 15% to 45%; adult incidence, 5% per year), with prevalence and incidence peaking between the ages of 35 and 55 years.4,5 As the world population gets older, low back pain is expected to increase substantially as an inevitable consequence of aging. Low back and neck pain in the United States was the third-largest condition of spending in 2013, with estimated health care spending of $87.6 billion.6 In recent years, there has been a substantial increase in health care costs to treat low back pain, and most of these treatments are simply to provide symptomatic relief with few strategies that actually contribute to correcting the underlying cause.7 Examples of these treatments are multiple but consistently embrace strategies that relieve symptoms before considering treatments designed to retard further degeneration. Lower back pain prevalence and social impact is further complicated by the growing levels of obesity and sedentary lifestyles, which are strongly associated with low back pain.8
Treatment for degenerative disc disease typically starts with nonsurgical methods that are intended to provide sufficient symptomatic relief by reducing pain from the damaged disc. Designated as conservative care, or nonsurgical management (NSM), methods of pain control may include anti-inflammatory medications, manual manipulation, steroid injections, electrical stimulation, back braces, and heat or ice therapy. In concert with treatments intended to provide symptomatic relief, other therapies such as physical therapy or yoga that restore functional mechanics are widely endorsed. Physical therapy provides an avenue for stretching and strengthening muscles that helps the lumbar spine heal and reduces the frequency of painful flare-ups. Several years ago, the North American Spine Society recommended postoperative physical therapy after surgery for degenerative conditions of the lumbar spine.9,10 Lifestyle modifications such as correcting posture, losing weight, or giving up smoking can sometimes help reduce stress on and improve healing of the damaged disc and can potentially slow down further degeneration.
In the end, structural degeneration changes loading and exacerbates both the mechanical and biologic-pathologic processes. These altered structural changes affect adjacent vertebrae and lead to facet joint arthritis, bone spur, or osteophyte formation and contribute to spine diseases such as canal stenosis and spondylolisthesis (Figure 1). The Thompson depiction of degeneration represents one of the first attempts to classify stages of disc degeneration, but many grading systems have been developed subsequently.11,12
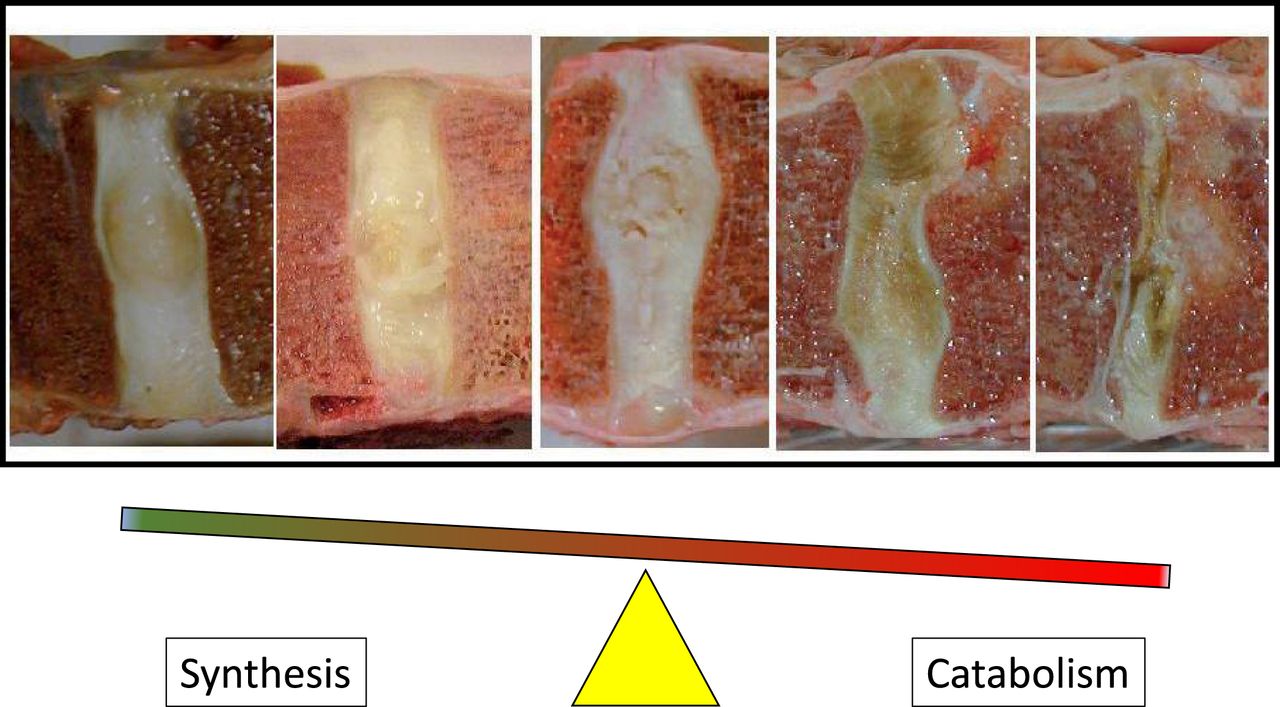
Disc degeneration is a progressive imbalance of anabolic and catabolic processes that result in tissue loss, which disrupts the anatomy, alters the loading, and results in progressive desiccation. Depiction of disc degeneration based on Thompson Grading.11
Intradiscal tissue loss resulting from disc degeneration is implicated as a factor in mechanical imbalance that, coupled with subsequent matrix loss, drives a cyclic process that gives rise to further instability and additional disc degeneration. This cycle of degeneration is repetitive and often progressive.
Whereas the exact origin of intervertebral disc degeneration remains to be identified, changes centered in the nucleus pulposus are believed to be the origin of the degenerative changes because the tissue in this region displays the severest change during the early stages of intervertebral disc degeneration.13 The variation in tissue composition during the degenerative process compromises the structural integrity, which further alters the metabolic activity and biochemical function within the disc.
The most common biochemical characteristics of intervertebral disc degeneration are the loss of glycosaminoglycan and the accompanying decrease of water content. These changes have been attributed to nutritional deprivation and inappropriate mechanical loading and are further linked to genetic, metabolic, and mechanical imbalances.14–20
How each of these causal factors leads to the various types of disc degeneration patterns and degeneration progression remains incompletely known despite the fact that variations in disc chemistry have been widely published and presented over the past 30 years.21 The critical remedy for intervertebral disc degeneration has yet to emerge as a structured solution for progressive disc degeneration.22–24 As the largest avascular structure in the human body, the intervertebral disc balances dual roles that require mechanical support without sacrificing spine flexibility. The uncertain origin of disc degeneration makes it difficult to determine whether matrix loss or mechanical perturbation is the primary insult to tissue homeostasis. The tissue integrity of the disc is further complicated by the lack of vascular, lymphatic, and direct neural integration directly to the central disc anatomy, and interventions intended for restoration require direct placement.
The development of chronic back pain in conjunction with degenerative disc disease often results in surgical treatment. Besides nonsurgical methods such as rest, physical therapy, and bracing, surgical procedures such as discectomy and placement of a total intervertebral disc prosthesis as well as replacement of the nucleus are now used for treatment of chronic back pain after disc herniation.25–31 So far, no clinically established procedure is available that slows the progression of intervertebral disc degeneration. In this context, the question has been raised about the ability of the intervertebral disc to regenerate its cellular matrix subsequent to either degenerative loss of tissue or discectomy.
Clearly, any reparative approach intended to facilitate motion preservation has to recognize that biomechanical stability and matrix composition are intrinsically connected. A strategy to retard or restore intervertebral disc tissue must initially stabilize the mechanical imbalance while at the same time supplement the disc matrix loss. The current unmet clinical need demonstrates that solutions to date fall short of achieving regenerative results and that beyond palliative relief, the clinical solution to the problem is essentially unsatisfactory.
Various biological techniques have been developed and tested to treat the degenerative intervertebral disc. Common to each has been the aim to sustain delivery of biologically active factors to the disc that might guide regeneration or at least retain the status quo of the affected tissue and retard further regeneration. General approaches that have been advance over many years include
Delivery of a growth factor, or other singular novel factor by intradiscal injection32,33
Gene therapy approaches that modify gene expression of resident disc cells in vivo (direct gene therapy)34,35
Autologous implantation of cells that have been antecedently removed, cultivated, and often modified in vitro36–41
Implantation of mesenchymal stem cells of a variety of in vitro–derived cell lineages has been proven in animal models.42–44
Using biologics heralds a new optimism that pain relief and functional improvement might be possible, but they lack attention to the structural deficit that results from disc degeneration. The structural deficit may be primary or secondary, but the difference is largely academic if the primary cause remains unknown. Supplementation of the disc addressing both biologic and tissue replenishment simultaneously might afford a distinct advantage in treating intervertebral disc degeneration.
This clinical study was developed to evaluate a technology that provides the benefits of allograft supplementation combined with a matrix replete with viable cells. Variations of porosity and cell density have been shown to affect the physical properties, such as the swelling ratio, stiffness, and mechanical strength, and theoretical reports have defined an optimal cell number that will produce the greatest disc repair.45,46 Replacing lost tissue or at least attempting to subsidize biomechanical components may overcome a loading imbalance resulting from tissue loss. Using a viable allograft matrix with cells that are anatomically appropriate for the spine is a second step, and being able to deliver the supplemental graft to the nucleus pulposus without causing undue additional damage to the annulus is the final necessary step.
The study was also conducted to evaluate whether a product developed for allograft supplementation could be safely administered and whether a formulation designed to replace tissue lost to degenerative disc changes might be presumably physiologic as well. There is no shortage of studies that have successfully transplanted cells into discs or nucleus pulposus material into disc, but no clinical applications that bring together the advantages of each have been tested.
MATERIALS AND METHODS
Clinical Study Design
The Viable Allograft Supplemented Disc Regeneration in the Treatment of Patients with Low Back Pain With or Without Disc Herniation (VAST) Trial was conducted, subjects enrolled, and data gathered under jurisdiction and oversight from the Sterling Institutional Review Board (Atlanta, GA) from February 2017 to August 2019. The subject evaluation has been extended to 24 and 36 months. The VAST Trial is registered at ClinicalTrials.gov (Identifier: NCT03709901).47
The VAST Trial was a prospective, randomized, parallel-arm, multicenter study approved to enroll up to 220 subjects at up to 15 clinical sites. Outcomes of the trial were based on assessment of primary and secondary endpoints 6 and 12 months after transplant of supplementary allograft compared with placebo or sustained conservative care (NSM) in subjects who have discogenic pain attributable to disc degeneration as judged by MRI scoring (Pfirrmann), physical examination, and subject-reported pain.
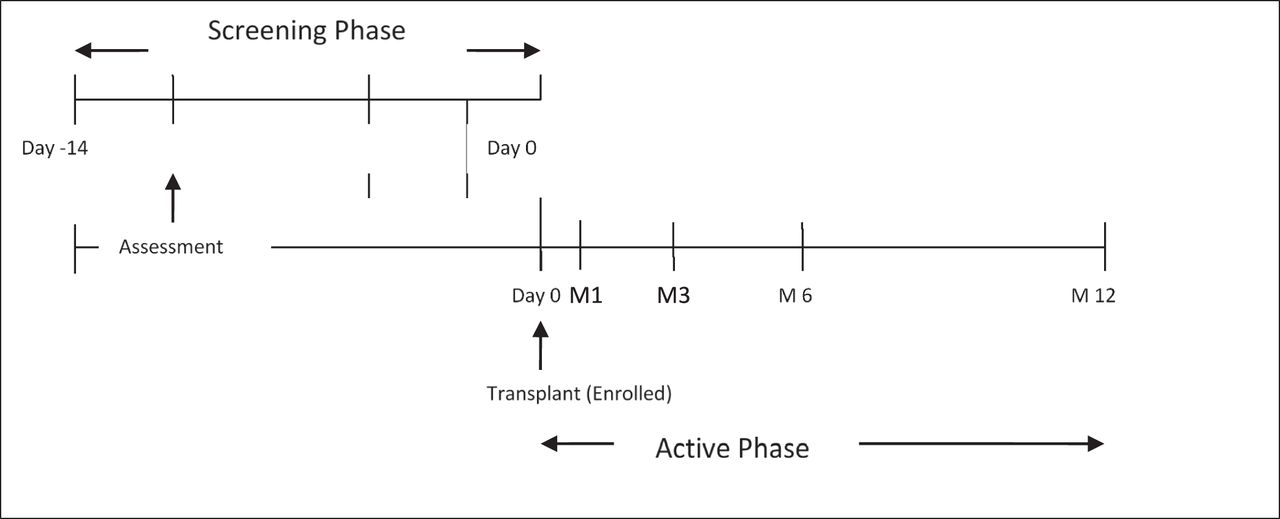
The study consisted of 2 phases: a screening phase (enrollment) followed by the active phase (12 months). As outlined (Figure 2), there was an indeterminate overlap of up to 2 weeks between the end of the screening phase and the start of the active phase for individual subjects. Subjects met entry criteria for both phases to be eligible and 2 weeks' opportunity was allowed for the subjects to evaluate the protocol and consent to their participation. Although 2 weeks were provided, subjects were eligible following their consent documentation being reviewed and received.

The study consisted of 2 phases: a screening phase (enrollment) followed by the active phase (12 months). As outlined, there was an indeterminate overlap of up to 2 weeks between the end of the screening phase and the start of the active phase for individual subjects. Subjects met entry criteria for both phases to be eligible and 2 weeks' opportunity was allowed for the subjects to evaluate the protocol and consent to their participation. Although 2 weeks were provided, subjects were eligible following their consent documentation being reviewed and received.
The aim of the VAST Trial was to investigate the clinical relevance of treating painful intervertebral disc tissue by a supplementary transplantation of viable cellular allograft disc matrix in a controlled clinical study comparing the cellular allograft with a saline placebo or continued treatment with NSM. Clinical differences among the 3 groups were compared by analyzing pain, functional restrictions, and neurological deficits using different scores that were especially developed for the assessment of degenerative spine complaints.48 In addition, radiographic evaluations and MRI were performed to evaluate morphologic changes that might distinguish differences between the 2 treatment groups. Radiographic evaluations and MRI included changes in the height of the intervertebral disc, Modic changes of the adjacent endplates, and the fluid content of the operated intervertebral disc.49,50 The target strategy of this new product is to slow down the complex degeneration process of the intervertebral disc and to offer a prescriptive option to therapeutic intervention that is clinically relevant and economically efficient.51
Subjects
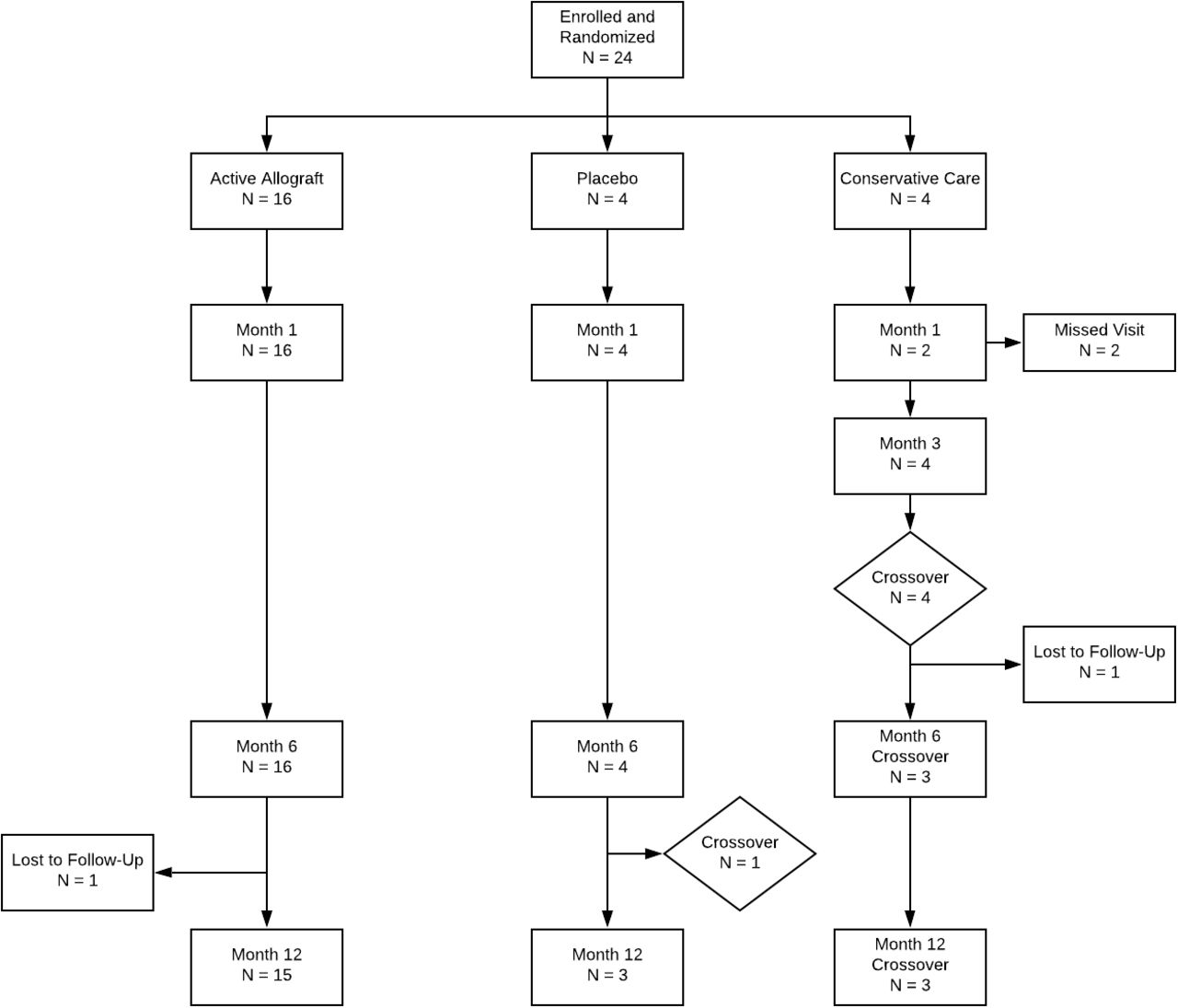
The study population in this report consisted of 24 subjects of various ethnicities from 18 to 60 years of age who demonstrated clinical disc degeneration at 1 or 2 vertebral levels from L1 to S1 (Figure 3). Patients were classified with moderate to severe disability (ODI ≥ 40%) and pain (VAS≥ 40 mm) that was chronic during the screening Phase and demonstrated modified Pfirrmann levels 3 to 6 on MRI. Subjects were included who demonstrated type 1 or type 2 Modic changes. Eligible subjects had experienced chronic low back pain for a minimum of 6 months, had no contraindications to allograft transplantation, and were willing to return to the clinic for multiple safety and efficacy assessments for up to 12 months following enrollment.
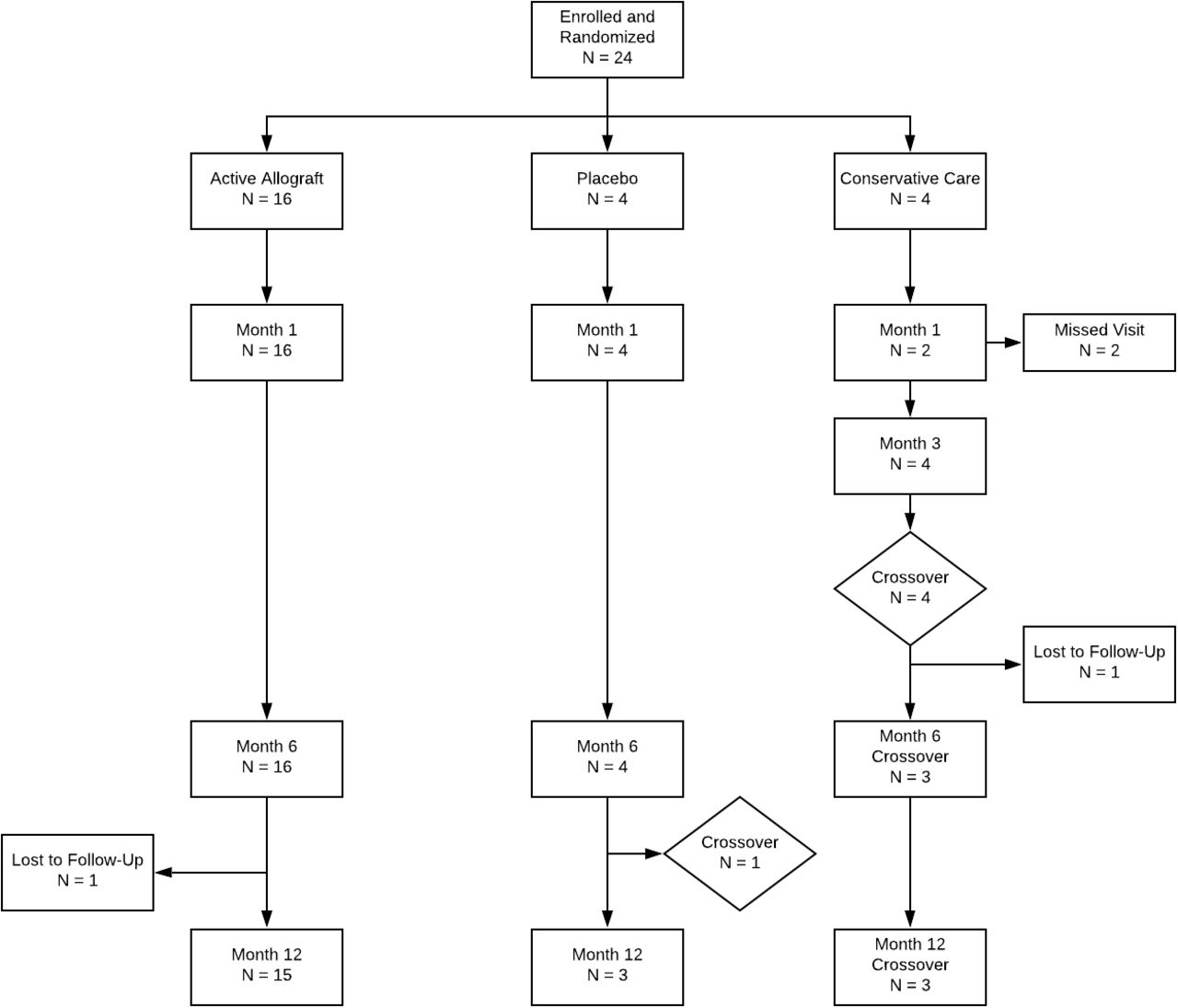
Subjects participating in the VAST Trial were randomized at 3.5:1:1 to receive a supplemental allograft (VIA Disc Matrix), saline as a placebo, or to continue under nonsurgical management (NSM). The assignment of the subjects to treatment (viable matrix supplement, saline placebo, or standard of care) was performed in a randomized manner after informed consent has been obtained.
Primary exclusion criteria included radicular pain, symptomatic spinal stenosis, disc protrusion >5 mm, spondylolisthesis >5 mm at any level, and body mass index >35. Detailed inclusion and exclusion criteria are presented in Table 1. Subjects participating in the VAST Trial were randomized at 3.5:1:1 to receive either a supplemental allograft (VIA Disc Matrix), saline as a placebo, or to continue under NSM. The assignment of the subjects to treatment (viable matrix supplement, saline placebo, or standard of care) was performed in a randomized manner after informed consent was obtained. Upon confirmation of eligibility, baseline measurements were collected and retained at individual study sites, and clinical data were monitored independently and entered by a third party (MileStone CRO, San Diego, CA).
Inclusion and exclusion criteria.
To evaluate and minimize subject risk, the first 24 subjects (at least 4 from each group) were evaluated 1 month after transplantation for a full safety assessment including radiographic and MRI information. All data collected were reviewed by the steering committee, who determined whether recruitment of patients as subjects could continue. The parameters used by the safety committee to determine whether recruitment resumed included the following:
A comparison of the adverse event (AE) profile between the 2 groups including rate, relationship, and severity.
A comparison of the clinical laboratory parameters between the 2 groups
A comparison of vital signs and physical examination findings between the 2 groups
A comparison of radiographic and MRI findings between the 2 groups
The first 24 subjects selected to participate in the study returned to the clinic for a safety assessment 1 month after their transplant. Subsequent clinic visits occurred at 6 and at 12 months following the transplant for safety and efficacy assessments. Subjects randomized to NSM were similarly followed to ensure no compromise in their care, and they additionally received a clinical follow-up by phone at 3 months to assess pain level and loss of function since their initial visit. Those subjects who felt they were not improving were offered VIA Disc Matrix and followed for an additional 12 months from the index procedure (implantation of the supplemental graft).
Study Interventions
Subjects screened, eligible, and randomized to the active phase of the study underwent a transplantation procedure. This procedure was performed under fluoroscopic guidance with moderate conscious sedation at the investigator's discretion in an outpatient setting. Subjects receiving the active treatment were treated identically to the point of the material preparation and the treatment placed. Subjects were not told of their treatment, and although a more detailed procedure for preparing the graft was required, subjects were not able to compare that preparation time with that of saline.
The preserved cell vial and mixing device were removed from the outer packaging using standard aseptic technique, presenting the cell vial and mixing device to the operative field. After the contents of the cell vial were completely thawed, the cell solution and nucleus pulposus allograft and saline components were mixed thoroughly.
Subjects were placed in the prone position on a multiplanar fluoroscopy table, propped, and bolstered for patient comfort and positioning for the procedure. Preliminary fluoroscopy was used to localize the target and mark the vertebral bodies and the disc(s) to be implanted. The skin was cleaned with betadine, and the subject was draped. Skin and deeper tissues were infiltrated with local anesthetic. A 22-gauge, 5-in spinal needle was inserted through Kambin's triangle at the index disc level(s) to be injected, care taken to avoid visceral, vascular, or neural injury. Placement of the needle was confirmed on additional imaging (anteroposterior and lateral fluoroscopic imaging, computed tomography [CT], CT fluoroscopy).
After determination that the needle tip was in an optimal location, the allograft was connected to the 22-gauge needle and 1.25 to 1.75 cm3 of the allograft was injected into the affected disc under real-time fluoroscopic imaging. Subjects were transferred to the recovery room for monitoring. The techniques for active transplant varied only in preparation of the material:
Supplemental viable allograft (VIA Disc Matrix) was prepared from human nucleus pulposus allograft that contains allogeneic viable cells. Each allogeneic product was individually prepared, but not specific for the subject being treated. A minimum of 6 × 106 cells were suspended in allograft matrix suspension.
Each subject who received the saline placebo was similarly positioned, the needle tip placement was identified, and the subject received approximately 1.75 cm3 per level as a placebo treatment.
Data Management
Data concerning the outcome of the supplemental transplant and the demographic data were collected during the visits and documented on appropriate case report forms. For all visits and for each patient, the case report forms were completed and all results uploaded by MileStone for data management and analysis after source data verification by the responsible monitor had been completed.
Interim Analysis
For safety and efficacy reasons, interim analyses performed after the first 24 subjects had completed the month 1 assessment of the active phase were done in a descriptive manner only and no unblinding occurred. A steering committee convened to review the results of the interim analysis and reviewed 6-month and 12-month data sets for VAS, ODI, and AEs.
Statistical Methods and Determination of Sample Size
The statistical evaluation was done with appropriate statistical software packages. For all baseline data, the descriptive statistics were presented. In the case of continuous parameters, statistical parameters for location and dispersion (eg, n, mean, standard deviation, range) were calculated. For discrete data, appropriate frequency tables were produced.
Primary Criteria
The primary criteria for the entire population were analyzed using a 2-sided approach on an α-level of .05 based on the total score. A hierarchical test procedure was applied. In the first step the pre-post difference of the groups was compared using the Wilcoxon rank sum test. When there was a significant result, a responder analysis was done; the analysis and results that followed spoke to the first 24-subject safety cohort but lacked adequate power for statistical analysis. Secondary variables were analyzed in an exploratory way using adequate statistical procedures to include the powered groups.
RESULTS
Efficacy and Safety Measurements
Per the study protocol, interim analysis of the first 24 subjects enrolled was undertaken at the 12-month follow-up, which mirrors the primary endpoint of the complete subject enrollment and analysis. An independent analysis and safety audit were carried out in conjunction with the data monitoring and review (Boston Biomedical Associates, Boston, MA). Demographics and baseline characteristics were similar among the 3 groups after randomization (Table 2).
Demographics and baseline characteristics safety cohort.
Efficacy and Safety Measurements
There were 204 subjects at 13 US sites enrolled in the VAST Trial. These individuals were segmented into a treatment group, an NSM group, and a saline placebo control group with a 3.5:1:1 randomization ratio. The first 24 participants were assessed at 1-month posttreatment to assess for safety. There were two coprimary endpoints including back pain as measured by the VAS and function as measured by the ODI. The primary endpoints were evaluated along with safety data and reported AEs and changes in clinical laboratory evaluations. The data were collected at baseline and at 3, 6, and 12 months. Structural evaluation was also performed and imaging studies including x-rays and MRI were performed at 6 and at 12 months.
Adverse events and serious AEs were evaluated in the 24-subject safety cohort. There were 13 events reported, 12 in the active allograft and 1 in the saline placebo. Events in the NSM cohort did not report any events associated with treatment even following crossover. Only 1 of the AEs was possibly associated with treatment. All events resolved, and none of the subjects left the trial. No serious AES were reported, and 6 of the 16 subjects accounted for the 12 AEs in the active allograft. Within the saline placebo group, 1 of the 4 reported an AE, and an additional subject who randomized to the saline placebo arm did not obtain meaningful pain relief and was provided the allograft at 6 months. In retrospect, 1 of 3 (33%) of the subjects randomized to placebo was affected, and 6 of 16 (37.5%) of those subjects randomized and blinded to the VIA Disc Matrix allograft were affected. The chief concern most often reported was back pain, and more than 50% of the AEs were generated from 3 of 16 subjects in the allograft cohort (Table 3).
Study cohort demographics.
Visual Analog Scale, Oswestry Disability Index
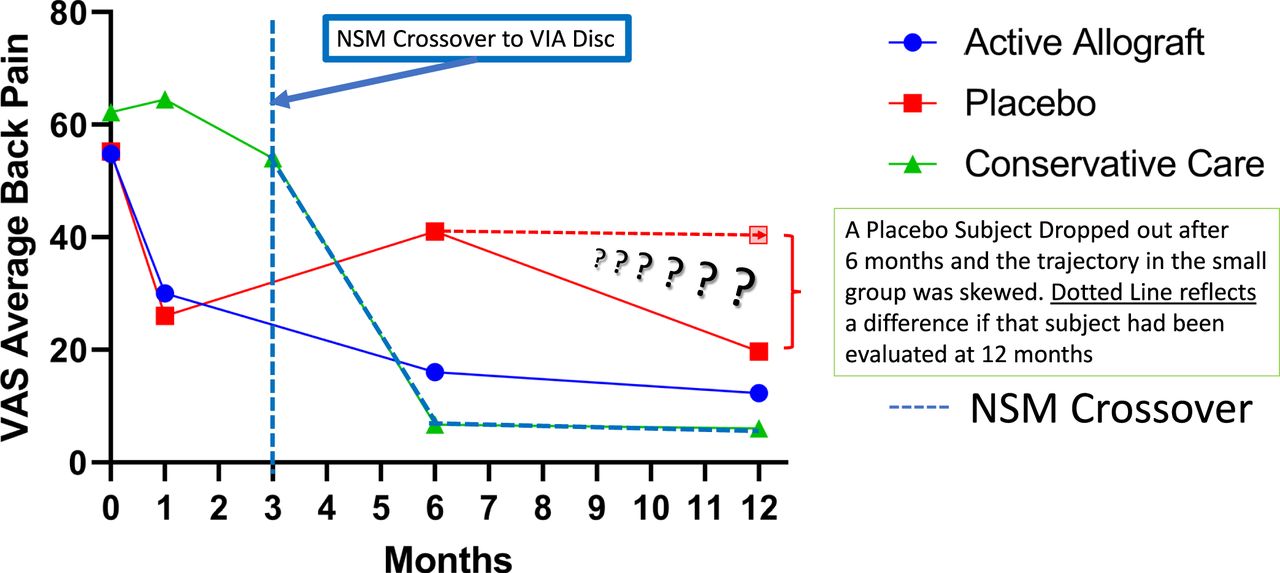
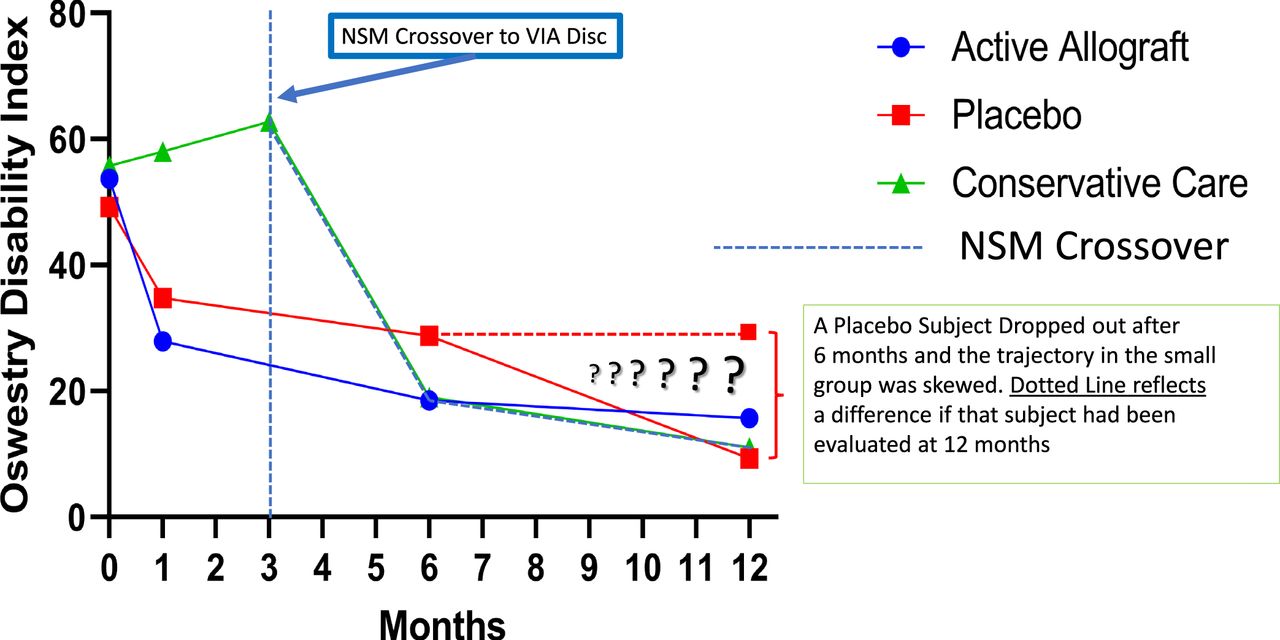
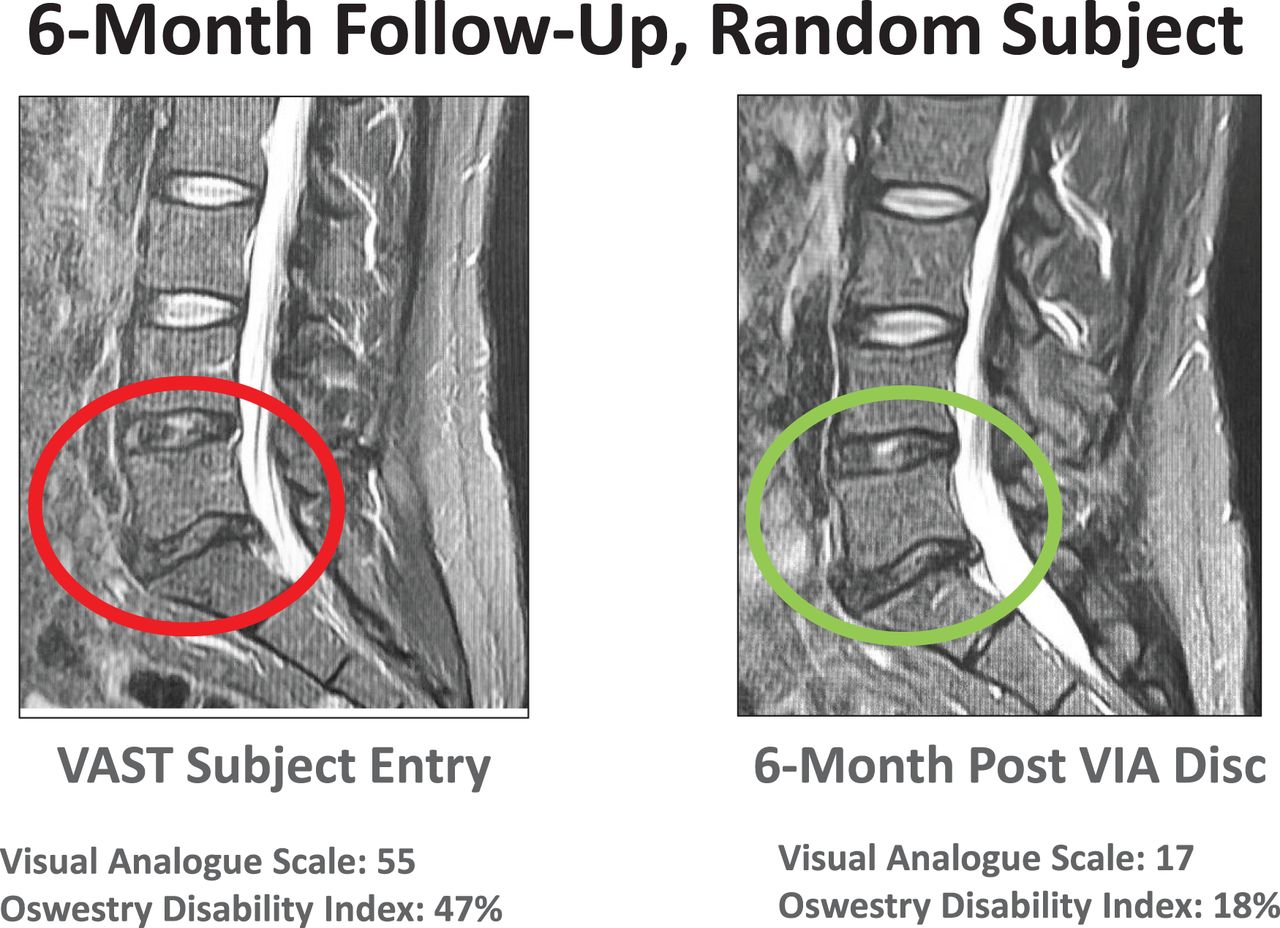
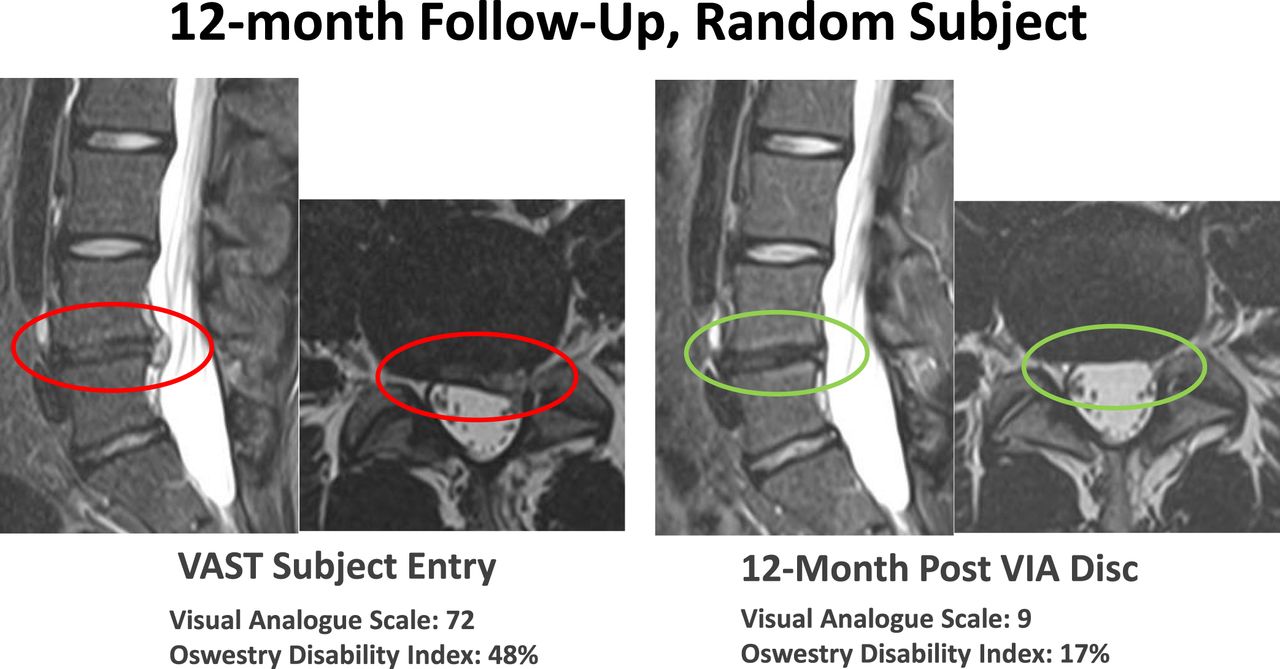
The data on the first 24 patients at 12-month follow-up were noted at the 6- and 12-month time points (Figures 4 and 5). The VAS back pain improved from 54.81, 55.25, and 62.25 in the allograft, placebo, and NSM subjects, respectively, to 16.0, 41.0, and 6.67 at 6 months and to 12.27, 19.67, and 6.0 at 12 months (Figure 4). At 3 months the VAS of the NSM group was 54.0. There was an option for the NSM patients to cross over to the allograft treatment group at the 3-month time point, and all subjects elected to cross over to allograft treatment. At the 6- and 12-month time points the ODI improved from 53.73, 49.25, and 55.75 in the allograft, placebo, and NSM subjects, respectively, to 18.47, 28.75, and 19.0 at 6 months, and to 15.67, 9.33, and 11.0 at 12 months (Figure 5). At 3 months the ODI of the NSM group was 62.75 and all subjects crossed over to allograft treatment. MRI evaluation showed anatomic improvement of the disc and enhanced nucleus signal (Figures 6 and 7).

Reduction in pain was evident in both of the active treatment groups in the study. Subjects who had been randomized to continue nonsurgical management (NSM) demonstrated no relief. The NSM subjects were further evaluated at 3 months (vertical dashed line), and unable to continue, all subjects crossed over to VIA Disc Tissue Matrix. Their response followed the trajectory of the active arms of the study and outcome demonstrated the largest improvement. One subject in the saline-treated group could not continue the study, and after 6 months crossed over to VIA Disc. The horizontal dashed line reflects that progress averaged into the small (4 subjects) placebo group. All patients receiving the randomized allograft completed the study in their cohort. In total, 21 of 24 subjects received the VIA Disc Tissue Matrix either randomized or open label.

Functional improvement was evident in both of the active treatment groups in the study. Subjects who had been randomized to continue nonsurgical (NSM) demonstrated no improvement. The NSM subjects were further evaluated at 3 months (vertical dashed line), and unable to continue, all subjects crossed over to VIA Disc Tissue Matrix. Their response followed the trajectory of the active arms of the study and outcome demonstrated the largest improvement. One subject in the saline-treated group could not continue the study, and after 6 months crossed over to VIA Disc. The horizontal dashed line reflects that progress averaged into the small (4 subjects) placebo group. All patients receiving the randomized allograft completed the study in their cohort. In total, 21 of 24 subjects received the VIA Disc Tissue Matrix either randomized or open label.
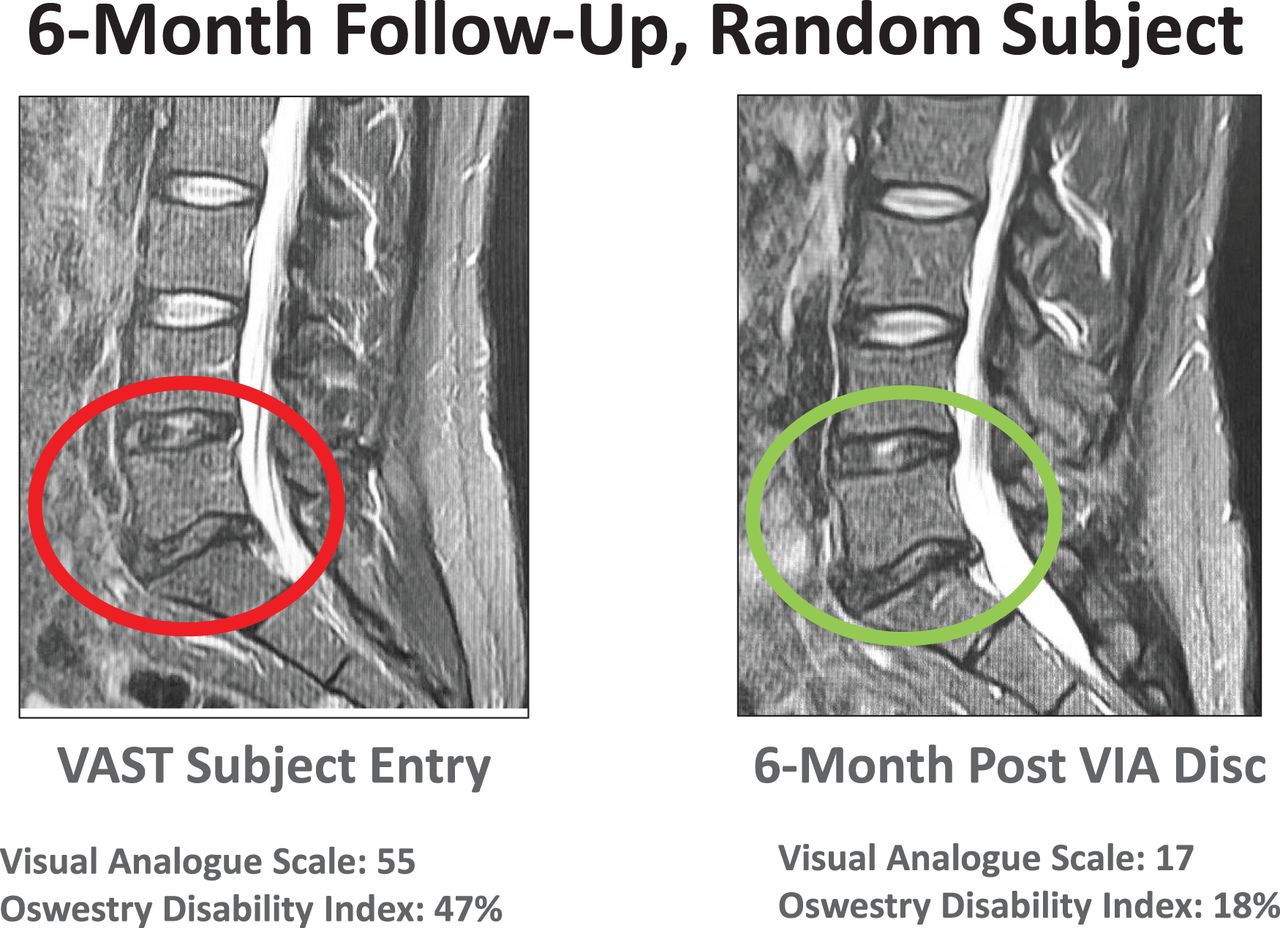
MRI images were used to qualify patients for inclusion in the study. Modified Pfirrmann scores between 3 and 6 were acceptable evaluations for participation. Signal intensity and morphologic distinction in the nucleus are noted. More important, pain reduction and functional improvement supported patient satisfaction in the treatment.

Images at 12 months demonstrated improvement in morphology, disc height, and patient indices of pain and functional improvement followed.
Data from the first 24 subjects as part of this large triple-arm prospective randomized control trial showed that a delivery of a viable structural allograft can be done safely with no AEs related to the procedures in these initial subjects followed up to 1 year. Subjects receiving the allograft had a larger reduction of pain and a greater functional improvement than did the placebo and NSM cohorts, and those NSM subjects crossing over to allograft supplementation attained similar pain and functional improvements to those initially randomized to receive the active treatment (Figures 4 and 5). The safety data were not powered for statistical significance, but the prominent improvements in pain and function trend toward the possibility of statistically significant differences at the final analysis of the data.
DISCUSSION
The high prevalence, incidence, and economic ramifications of degenerative disc disease have guided numerous attempts to intervene with treatments that provide more than palliative solutions to pain. All previous and current human studies of transplanted cells have involved the use of autologous or allogeneic cells requiring ex vivo expansion of the cells. This expansion is costly, time-consuming, and strictly regulated, guiding technology that to date has not provided compelling data for clinical translation. Using allograft preparation technology that has been sufficiently refined to produce allograft that has been minimally manipulated, this study has demonstrated that it is possible to safely offer percutaneous delivery that reduces pain, enhances functional recovery, maintains disc height, and can be administered in a physician's office using sterile technique.
Several key observations emerged from the interim analysis of the VAST Trial. Although conducted with a small population, the 12-month trial provided encouraging evaluations:
Adverse events were limited to a small subpopulation within the study and were transitory and not related to the treatments. All events resolved, although a couple of subjects required treatment. The percentages of subjects in the trial were approximately similar, but the small numbers in this safety cohort may not be applicable to larger treatment populations.
Supplemental allograft appears to provide an effective resolution of pain, to enhance functional recovery, and to sustain disc height not only at the index level but at levels rostral to the treated level(s) as well.
All 16 subjects randomized to and receiving the allografts showed progressive and sustained improvement over the 12 months over which data were collected.
Within the cohort randomized to receive saline placebo, 1 of the 4 subjects did not improve and, beset by unremitting pain, crossed over and was treated with VIA Disc Matrix in an open-label manner.
None of the subjects randomized to NSM were able to complete the study in their cohort. All 4 subjects crossed over to receive the VIA Disc Matrix after an additional 3 months of NSM and demonstrated immediate and marked improvement in both pain relief and functional improvement that was sustained through the 12-month evaluation.
Meaningful clinical improvement was achieved in the cohort randomized to receive VIA Disc Matrix by 6 months and was maintained over the course of the study, attaining a 71% reduction at 6 months that reached 78% by 12 months. Those subjects receiving placebo were only able to achieve a 26% reduction at 6 months. Conservative-care subjects crossed over to open-label VIA Disc at 3 months, and by 6 months had attained 88% reduction in pain, which at 12 months was 89%. Note that 1 of the subjects receiving the placebo could not continue to 12 months, and the remaining 3 did achieve 65% reduction but from a population reduced and selected by the loss of a subject due to intolerable pain.
Meaningful clinical improvement as measured by ODI was achievable by 6 months and maintained over the course of the study. Subjects randomized to VIA Disc achieved 35-point improvement by 6 months that improved by 45.67 points at 12 months. The placebo group achieved a 20.51-point improvement at 6 months and, despite the dropout of 1 subject, achieved 39.92 points at 12 months. Subjects originally randomized to continue conservative care did not fare well until crossing over and following the open-label VIA Disc placement, achieved 43.35-point improvement by 6 months that further improved to 51.75 points by 12 months.
Subjects who crossed over and were aware of receiving the supplemental allograft demonstrated the largest net gains and the fewest AEs in the study.
The critical component of safety was demonstrated in this 24-subject interim analysis of the VAST Trial data. Separate from safety, the enrolled subjects demonstrated meaningful clinical improvement and disc height sustenance. The supplementation of the disc with viable allograft was able to achieve a marked reduction in pain, an improvement of function, and a resolution of symptoms in degenerated discs detect by MR imaging, which was encouraging. The context and connection of image and symptoms has been defined several times, notably demonstrating that imaging alone is an insufficient index to evaluate clinical symptoms.52,53
Given the durability of the allograft over the 12 months of the study, it will be interesting to evaluate the data from the larger group to see whether the results from the first 24 patients are representative of the entire population studied. The key comparison will be in the improvement noted in the symptomatic discs, which differs significantly from imaging studies that evaluate disc degeneration in asymptomatic patients. Being able to show a comparable improvement in anatomical evaluation in concert with the remission of pain offers unique information that could help to identify the appropriate patients who may benefit from the treatment. Treatments that avoid care or mask symptoms without improving the condition do little for the patient or the health treatment community. By using a supplemental allograft that retains viable components of healthy disc, this treatment combines the expectation of mechanical support with biochemical process to promote disc healing while providing structural support. Being able to reduce disability, enhance activity, and partially preserve spinal motion provides a meaningful clinical intervention that has been shown to be safe and, in this small population, effective.
Whereas the mechanism of action remains to be fully elucidated, many studies have suggested that a variety of viable cells transplanted into the disc can generate extracellular matrix and provide the necessary cellular components required to prevent further disc degeneration following discectomy. As noted in the introduction, cell therapy alone has not shown itself to provide sufficient intervention with regard to either efficacy or duration. The opportunity to pair a disc tissue matrix with cells may provide a supplemental allograft to supplement the lost tissue and cells consequent to the degenerative process. Although it is impossible to fully know whether the cells can differentiate and survive or produce signaling agents that cause the intervertebral disc to heal, this study does confirm that this allograft technology can be administered in a controlled, safe, and regulated environment and that it has produced positive provisional clinical results.
Footnotes
Disclosures and COI: Dr Beall reports grants from Medtronic, nonfinancial support from Amendia, other from Lilly, other from Synthes, other from Johnson and Johnson, grants from Vitacare, grants from Ortho Kinematics, other from DFine, other from Bone Support, other from Convatec, other from Spinal Ventures, grants from Zyga, grants from Liventa, grants, nonfinancial support and other from Vexim, grants from Mesoblast, grants, personal fees, nonfinancial support and other from Vivex, outside the submitted work.
- ©International Society for the Advancement of Spine Surgery
- This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery. Copyright © 2020 ISASS.
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.