


ABSTRACT
Background: Expandable devices for transforaminal or posterior lumbar interbody fusion (TLIF and PLIF, respectively) may enable greater restoration of disc height, foraminal height, and stability within the interbody space than static spacers. Medial-lateral expansion may also increase stability and resistance to subsidence. This study evaluates the clinical and radiographic outcomes from early experience with a bidirectional expandable device.
Methods: This was a retrospective analysis of a continuous series of patients across 3 sites who had previously undergone TLIF or PLIF surgery with a bidirectional expandable interbody fusion device (FlareHawk, Integrity Implants, Inc) at 1 or 2 contiguous levels between L2 and S1. Outcomes included the Oswestry Disability Index (ODI), a visual analog scale (VAS) for back pain or leg pain, radiographic fusion by 1 year of follow-up, subsidence, device migration, and adverse events (AE).
Results: There were 58 eligible patients with radiographs for 1-year fusion assessments and 45 patients with ODI, VAS back pain, or VAS leg pain data at baseline and a mean follow-up of 4.5 months. The ODI, VAS back pain, and VAS leg pain scores improved significantly from baseline to final follow-up, with mean improvements of 14.6 ± 19.1, 3.4 ± 2.6, and 3.9 ± 3.4 points (P < .001 for each), respectively. In addition, 58% of patients achieved clinically significant improvements in ODI, 76% in VAS back pain, and 71% in VAS leg pain. By 1 year, 96.6% of patients and 97.4% of levels were considered fused. There were zero cases of device subsidence and 1 case of device migration (1.7%). There were zero device-related AEs, 1 intraoperative dural tear, and 3 subsequent surgical interventions.
Conclusions: The fusion rate, improvements in patient-reported outcomes, and the AEs observed are consistent with those of other devices. The bidirectional expansion mechanism may provide other important clinical value, but further studies will be required to elucidate the unique advantages.
Level of Evidence: 4.
INTRODUCTION
Lumbar interbody fusions, through a posterior (PLIF), transforaminal (TLIF), lateral (LLIF), or anterior (ALIF) approach, are routinely performed to treat a variety of spinal pathologies.1–3 PLIF and TLIF are the most commonly performed procedures for lumbar fusions. These procedures use a posterior approach to the disc space through a narrow window, the Kambin triangle, created by partial or complete laminectomy and facetectomy. PLIF and TLIF allow for both direct and indirect neural decompression as well as posterior supplemental fixation through a single approach. However, the geometry and size of PLIF and TLIF interbody fusion devices are limited by the narrow access window available to deliver the device.4 Much larger spacers can be implanted through LLIF or ALIF techniques, allowing for better endplate coverage with fewer constraints on restoring disc space height and lordotic angle. However, these techniques are dependent on indirect decompression, generally increase the risk to visceral and vascular structures, often involve at least 2 positions or incisions for supplemental fixation, and may require an exposure surgeon. The smaller PLIF and TLIF spacer size may increase the risk of subsidence or migration4,5 and limit the restoration of disc and foraminal height compared with ALIF or LLIF spacers.6,7
A variety of expandable interbody fusion devices have been introduced to help overcome the geometry and size constraints traditionally associated with PLIF and TLIF devices.4 These expandable devices are inserted with a small initial profile and expanded in situ to increase the device's footprint, typically in height (cranial-caudal plane), allowing for increased lordotic angle by lengthening the anterior column. Medial-lateral expansion may help to resist subsidence, whereas cranial-caudal expansion enables greater restoration of disc height and foraminal height than static (nonexpandable) spacers.8,9 In situ device expansion may improve endplate contact while minimizing insertion forces and potential endplate damage,10 which may help reduce the risk of spacer migration or subsidence.11 These expandable designs are compatible with minimally invasive surgical techniques, which are well established and continue to gain popularity in spinal surgery. Most expandable spacers use complex articulation mechanisms to expand the implant profile in 1 plane, either cranial-caudal (height) or medial-lateral (width), but typically do not achieve biplanar expansion. Frequently, these complex mechanisms decrease graft chamber dimensions, limiting the space available for bone grafting and, rarely, may predispose the spacer to mechanical failure.
The current study evaluated the safety, clinical outcomes, and radiographic characteristics of a novel expandable implant (FlareHawk, Integrity Implants, Inc, Palm Beach Gardens, FL) that achieves bidirectional expansion through a relatively simple mechanism. The current study retrospectively reviewed a continuous series of patients treated with a novel, bidirectional expandable implant across 3 sites to evaluate the clinical safety and effectiveness as well as radiographic fusion success.
METHODS
Patient Population
Patients who underwent TLIF or PLIF surgery using a bidirectional expandable spacer (FlareHawk Expandable Interbody Fusion System, Integrity Implants, Inc) before September 30, 2018, were included in this study. Surgeons from 3 sites performed a retrospective chart review using the eligibility criteria of the US Food and Drug Administration (FDA)–cleared product labeling (Table 1). A central institutional review board (Western Institutional Review Board, Puyallup, WA) approved the study protocol and investigators prior to study commencement and granted a waiver for patient informed consent, considering that the research was limited to retrospective chart review.
Study Eligibility Criteria
Bidirectional Expandable Interbody Fusion Device
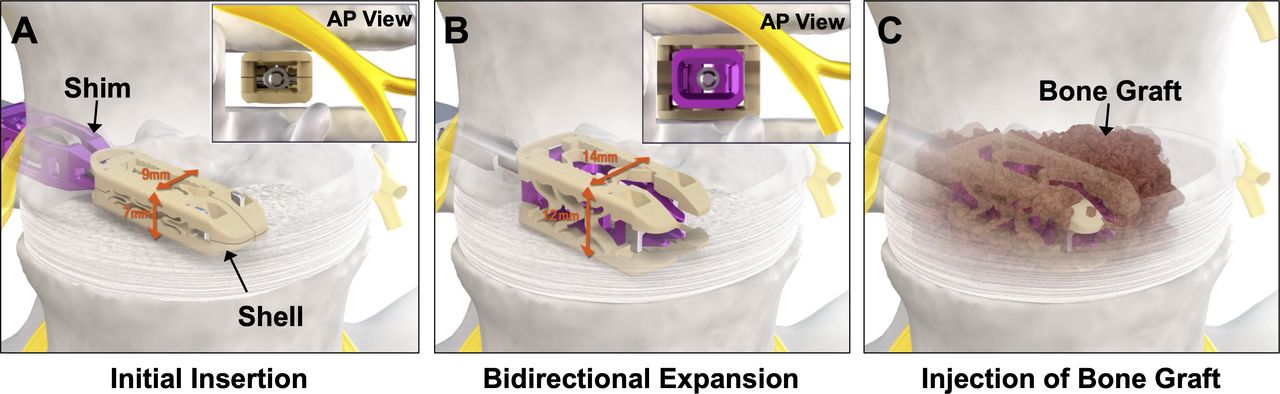
The bidirectional expandable device consists of a polyether ether ketone (PEEK) shell and a titanium shim component. The PEEK shell component is a rectangular frame with flexible struts on all 4 sides that allows insertion into the intervertebral body space in a nonexpanded form (Figure 1A). The shell contains an integrated titanium core that anchors the inserter during delivery of the shim. The shim has a tapered front end that inserts into the shell component and bidirectionally expands the shell within the intervertebral body space. When fully inserted, the shim locks to the core within the shell to provide structural stability for interbody fusion (Figure 1B). The implant is then backfilled with bone graft, which is delivered through the device and distributed into the intervertebral body space via the open device design (Figure 1C).

Illustration of (A) initial insertion, (B) bidirectional expansion, and (C) injection of bone graft.
Surgical Technique
TLIF or PLIF surgery was performed as described previously, using either an open or minimally invasive approach.12–14 Following exposure, decompression and preparation of the disc space was performed, including sequential distraction. Implant sizing was determined using trials and/or evaluation of preoperative and intraoperative imaging. The shell and shim were loaded inline onto an inserter instrument, and the shell was placed into the interbody space (Figure 1A). The shim was then advanced into the shell to achieve in situ expansion of the interbody space (Figure 1B). Locking of the 2 components was verified through direct visualization and/or intraoperative x-ray. Autologous and/or allogenic bone graft was packed into the device and disc space through a funnel impactor (Figure 1C). Supplemental pedicle screw/rod fixation was used in all cases.
Demographic and Intraoperative Data
Patient demographic data and intraoperative data were collected from the electronic health records. Baseline demographic data included age, gender, body mass index (BMI), smoking status, diagnoses, and any comorbid conditions, such as diabetes. Diagnoses were collected and summarized based on International Classification of Diseases, Tenth Revision (ICD-10) codes. Intraoperative data included the surgical level(s), surgical approach, estimated blood loss, length of surgery, length of hospitalization, and any intraoperative adverse events (AE).
Clinical Outcomes
Patient-reported clinical outcome measures included a visual analog scale (VAS) for back pain, VAS for leg pain, and the Oswestry Disability Index (ODI). The VAS score for back and leg pain was anchored by no pain (score of 0) and pain as bad as it could be or worst imaginable pain (score of 10). The ODI was used to quantify disability related to lower back pain. The score ranged from 0 to 100. A score of 0 to 20 reflected minimal disability; 21 to 40, moderate disability; 41 to 60, severe disability; 61 to 80, crippled; and 81 to 100, bedbound. Patient-reported outcome data were collected from the last available follow-up in which the questionnaires were completed.
Interbody Fusion Assessment
Radiographic fusion was assessed according to the Bridwell-Lenke grading system15 using planar radiographs collected at 12 ± 3 months follow-up. Grades 1 or 2 were accepted as fused and grades 3 or 4 were accepted as not fused. All radiographs were evaluated by 2 independent readers (1 neurosurgeon and 1 radiologist). A third independent reader (neurosurgeon) served as an adjudicator for any discrepancies between the 2 primary readers regarding fusion status. Radiographs were also evaluated for cage migration or subsidence. Migration was defined as a displacement of the device relative to the position within intraoperative or immediately postoperative images. Subsidence was defined as an overlap between the vertebral endplates and the device exceeding 25% of the device height.16
Adverse Events
All intraoperative AEs including device component breakage/fracture, device malfunction, dural tear/injury, nerve root injury, device-related delay of surgery, retained foreign body, and others were recorded. Postoperative AEs including infection, donor site pain, complex regional pain syndrome, urinary retention, ipsilateral or contralateral radiculopathy, device- component fracture, migration/retropulsion, subsidence, complication (eg, loss of expansion), loss of fixation, nonunion, vertebral fracture, adjacent segment disease, and others were assessed by each investigator in terms of relatedness to the device or surgical procedure. The intensity of each AE was based on the definitions established by the World Health Organization (mild, moderate, severe, or death). Device and procedure relatedness were prospectively defined in the protocol prior to initiating the chart review.
Statistical Methods
Descriptive statistics including means, standard deviations, medians, ranges, frequencies, and/or 95% confidence intervals (CI) were calculated for each variable using R software, Version 3.6.0 (R Foundation for Statistical Computing, Vienna, Austria; http://www.R-project.org/). Paired t tests or paired Wilcoxon signed rank tests were used to determine whether changes in VAS or ODI scores were significantly different from preoperative baseline to follow-up. Significant clinical improvements were based on minimum clinically important differences (MCID) of at least 10 points (100-point scale) for ODI and at least 2 points (10-point scale) for VAS leg or back pain.17 Interrater agreement in fusion grade assignment was assessed through percentage agreement, Cohen κ, as well as prevalence- and bias-adjusted κ. Subgroup analyses were performed using multiple logistic regression, χ2 and/or Fisher exact test to determine whether there were any significant differences in outcomes on the basis of demographic or operative factors. Statistical significance was accepted for P < .05.
RESULTS
Data Availability
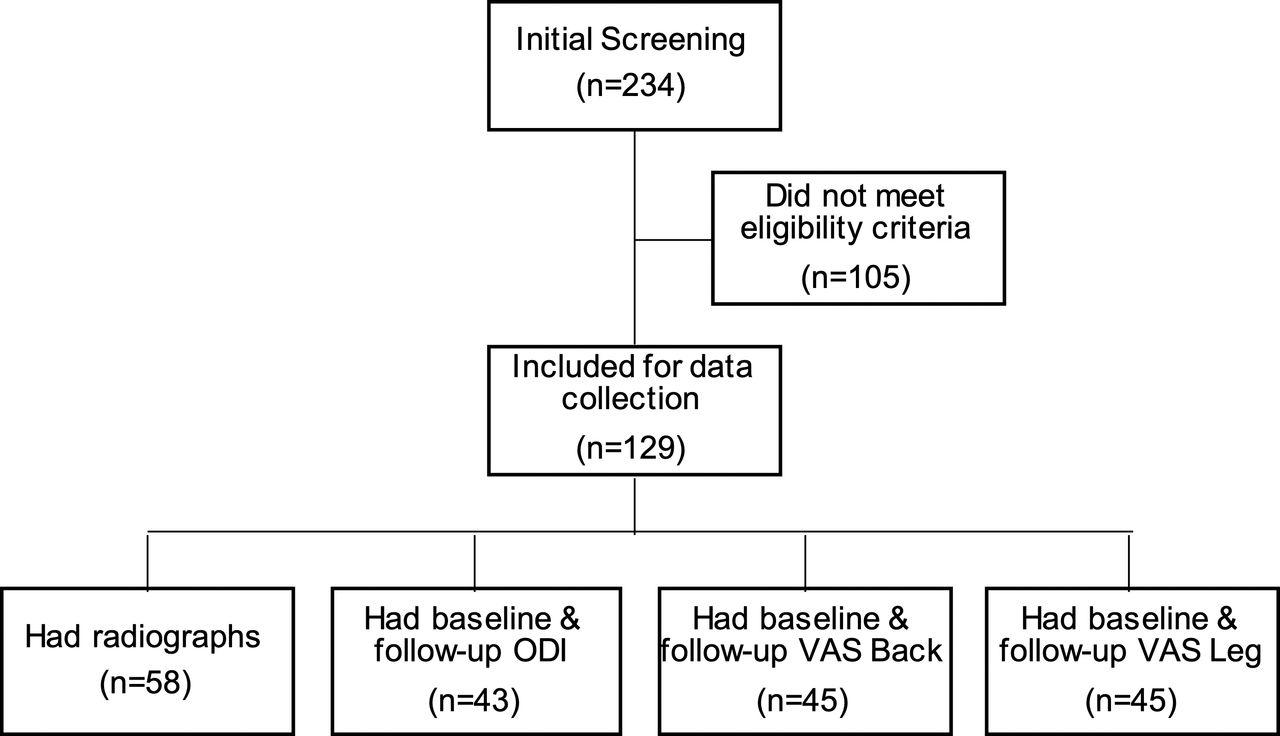
A total of 129 patients met all eligibility criteria, having undergone surgery between February 2017 and September 2018 (Figure 2). There were 58 patients evaluable for radiographic fusion at 1 year follow-up. Forty-three study patients had ODI data at both baseline and follow-up, with an average follow-up of 4.4 ± 3.8 months (range, 0.5–20.5 months). For VAS leg pain, 45 patients had data at both baseline and follow-up, with an average follow-up of 4.6 ± 4.4 months (range, 0.5–20.5 months). Scores for VAS back pain were available for 45 patients, with an average follow-up of 4.4 ± 4.3 months (range, 0.5–20.5 months).
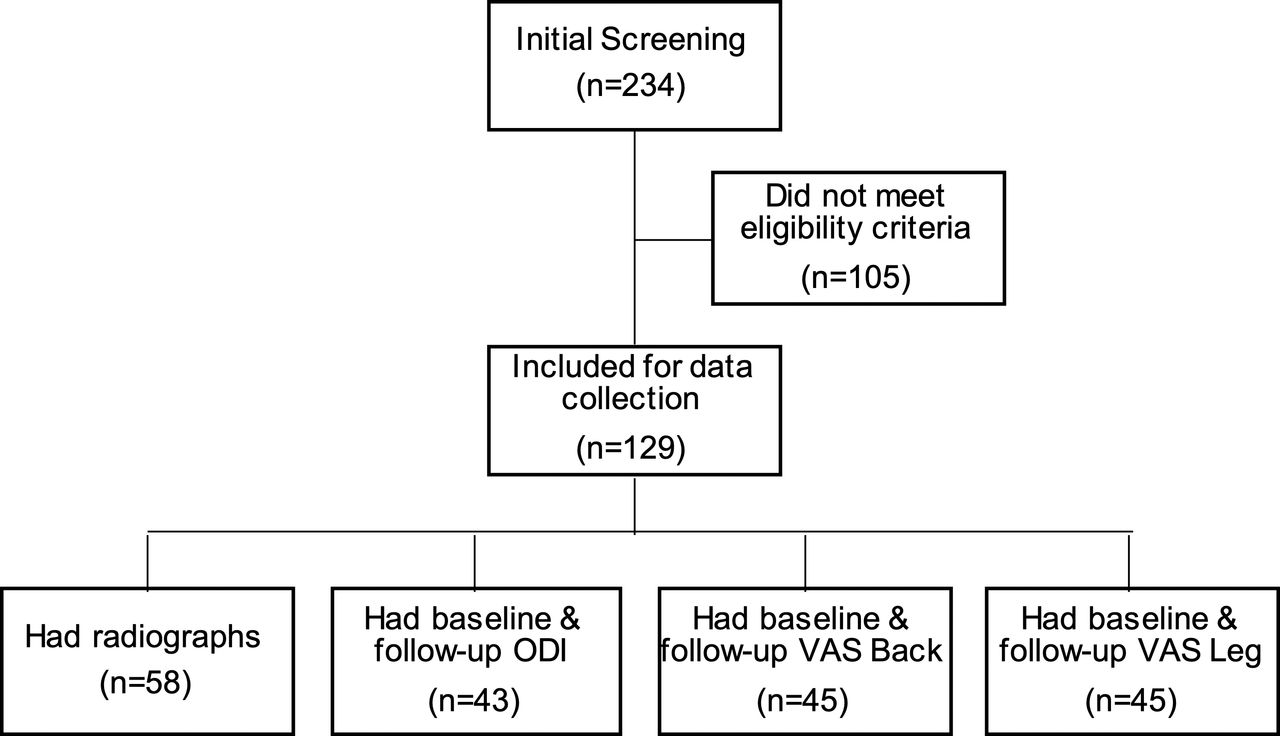
Flowchart of subject screening and follow-up data availability.
Baseline, Demographic, and Operative Data
The mean age of participants was 60.5 ± 12.7 years (range, 31.8–86.8 years), and 56% were women. The mean BMI was 31.3 ± 7.3 (range, 16.7–55.8) and 47% of patients were considered obese (BMI ≥ 30). Furthermore, 20% of patients were active smokers and 22% had a history of smoking. Most study patients had medical comorbidities including obesity (47%), hypertension (41%), diabetes (19%), cancer or history of cancer (10%), coronary heart disease (7%), hypercholesterolemia (6%), and/or hypothyroidism (5%). The most frequent preoperative diagnoses were spinal stenosis (67%), spondylolisthesis (51%), radiculopathy (27%), other disc degeneration (25%), low back pain (5%), and spondylosis (5%).
Among the 129 study patients, TLIF (51%) or PLIF (49%) surgery was performed on 171 levels. The most common surgical level was L4-L5 (55%) followed by L5-S1 (28%), with 65% of patients undergoing a 1-level fusion procedure and 35% undergoing a 2-level procedure. A minimally invasive approach was used in 88% of cases, and the remaining 12% underwent open procedures. Allograft alone was used in 26% of patients. Autograft alone was used in only 6% of patients, whereas both autograft and allograft were applied in the remaining 68% of patients. Unilateral posterior supplemental fixation was used in 24% of patients versus bilateral pedicle screw constructs in the other 76% of patients. The mean surgical time was 175 ± 54 minutes (range, 77–400 minutes) and the mean hospital length of stay (LOS) was 1.8 ± 1.9 days (1 day LOS: 69%, 2 days LOS: 16%, >2 days LOS: 15%; Table 2).
Summary of Operative Procedure Variables
Patient-Reported Outcomes
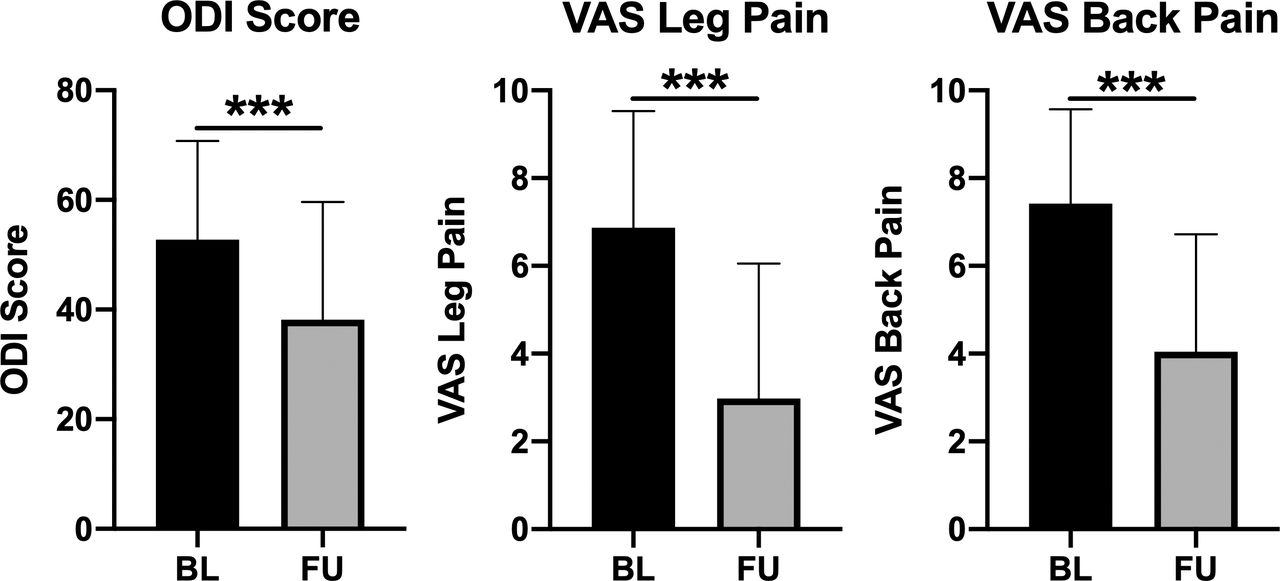
On average, there were significant improvements from baseline to follow-up in ODI, VAS leg pain, and VAS back pain (P < .001 for each; Figure 3). The average improvements were 14.6 ± 19.1 points in ODI (100-point scale), 3.9 ± 3.4 points in VAS leg pain (10-point scale), and 3.4 ± 2.6 points in VAS back pain (10-point scale). The proportion of patients achieving a clinically significant improvement on the basis of the MCID for ODI, VAS leg pain, or VAS back pain was 58%, 71%, and 76%, respectively. There were no significant differences in ODI, VAS leg pain, or VAS back pain outcomes on the basis of the number of levels treated (1 level vs 2 levels) or the type of supplemental fixation (unilateral vs bilateral).

Patient-reported outcomes, including the Oswestry Disability Index (ODI), visual analog scale (VAS) for leg pain, and VAS for back pain were significantly improved (decreased) from baseline (BL) to last follow-up (FU). *** indicates P < .001 based on Wilcoxon matched-pairs signed rank test.
Radiographic Assessments
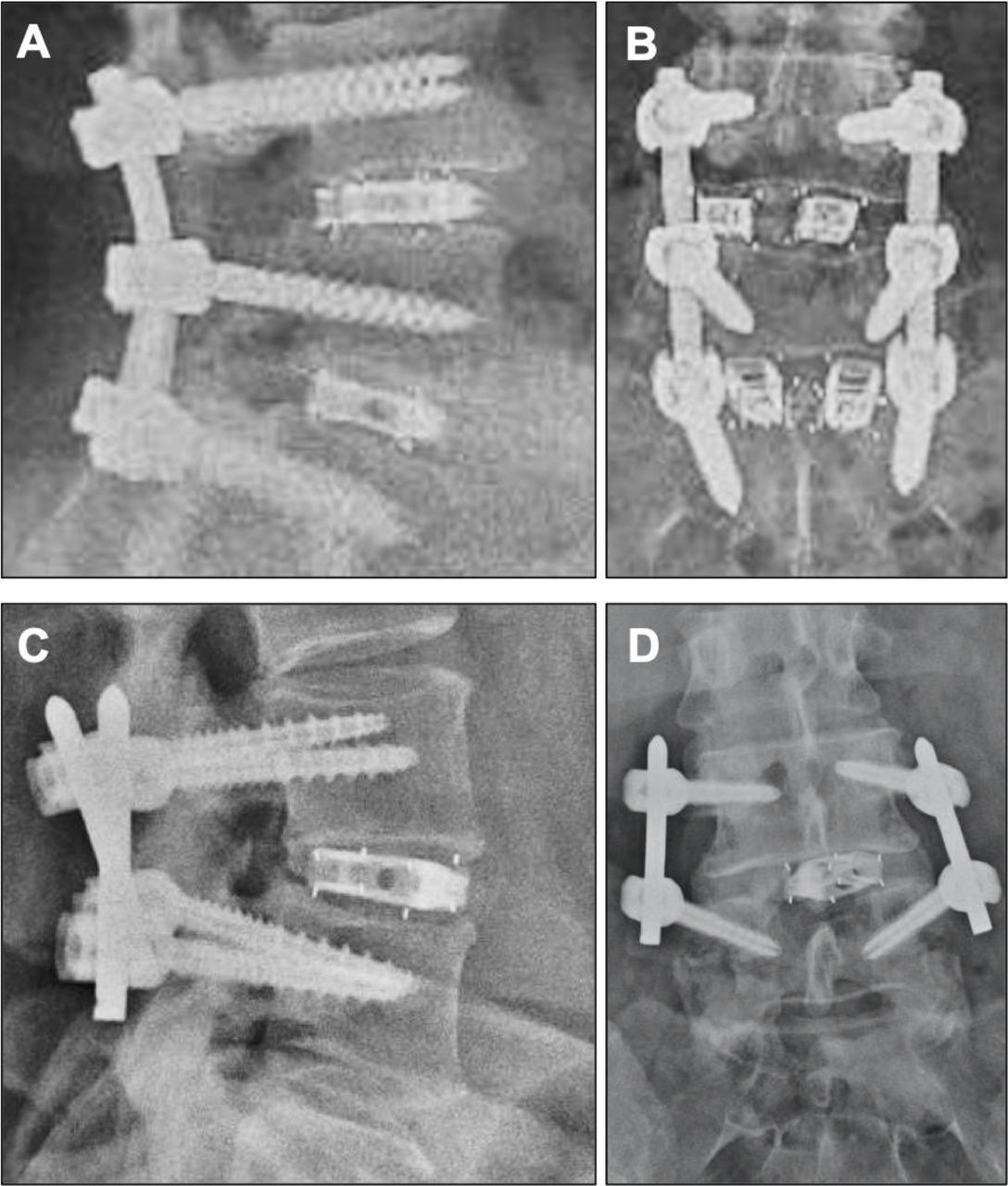
Among the 58 patients (77 levels) evaluable for radiographic fusion at 1 year follow-up, 56 patients (96.6%; 95% CI: 88.3%–99.0%) and 75 levels (97.4%; 95% CI: 91.0%–99.3%) had achieved fusion based on the Bridwell-Lenke grading system (Figure 4). One patient who was determined to not be fused had a 2-level procedure, with 1 level graded as fused and 1 level not fused. Both nonfused patients were active or previous smokers. No significant differences were observed in the fusion success rate on the basis of the number of levels involved (1 level [n = 39]: 97.4%, 2 level [n = 18]: 94.4%; P = .54) or the type of bone graft used (allo/auto [n = 28]: 93%; allo [n = 22]: 100%; auto [n = 8]: 100%; P = .33). There were zero cases of subsidence exceeding 25% of the device height and 1 case of device migration. There was 1 reviewer discrepancy regarding a fused versus nonfused grade, which was adjudicated by a third reviewer as not fused. The percentage agreement in Bridwell-Lenke grades for each fusion level was 81.8% between the 2 primary reviewers, and Cohen κ was 0.62 (95% CI: 0.45–0.79), indicating substantial agreement. The percentage agreement in fusion status was 98.7% between the 2 primary reviewers, with a prevalence- and bias-adjusted κ of 0.97, indicating very good agreement.

Examples of fusion in (A, B) a 2-level TLIF procedure and (C, D) a 1-level PLIF procedure. TLIF, transforaminal lumbar interbody fusion; PLIF, posterior lumbar interbody fusion.
Complications and Additional Surgeries
There was 1 intraoperative AE (0.8%; 95% CI: 0.1%–4.3%), consisting of a dural tear that was resolved at the time of surgery without nerve root injury. The dural tear was not considered to be device related. The patient with an intraoperative dural tear also reported weakness in the right leg at approximately 10.5 months follow-up, but the 2 events were not clearly related. Two patients (1.6%) had postoperative AEs adjudicated as severe, which consisted of radiculopathy and bilateral leg pain deemed unrelated to the interbody device. In the patient with bilateral leg pain, failed supplemental instrumentation was observed during the revision surgery at approximately 4 months after the index procedure. There were zero interbody device-related AEs (95% CI: 0%–2.9%) and 12 procedure-related AEs in 9 patients (7.0%; 95% CI: 3.7%–12.7%). Procedure-related AEs consisted of adjacent segment disease at 14 months postoperatively (n = 1), nonunion and failed supplemental instrumentation at 20 months (n = 1), failed supplemental instrumentation at 3.5 months (n = 1), radiculopathy (n = 2), lucencies around screws (n = 2), superficial infection (n = 2), and postoperative pain, discomfort, or swelling (n = 3).
There were 3 subsequent surgical interventions in 3 patients (2.3%; 95% CI: 0.8%–6.6%). One patient developed symptomatic adjacent segment disease and underwent TLIF surgery at 2 additional levels. This patient was obese (BMI > 45) and an active smoker. The patient with a nonunion and failed supplemental instrumentation at L5-S1 underwent revision surgery at 20 months and previously had a spinal cord stimulator implanted at 6.5 months after the index procedure. The subject was an active smoker with additional comorbidities including hypertension and coronary heart disease. The third subject experienced severe bilateral leg pain approximately 3 months postoperatively, and the investigator observed failed supplemental instrumentation upon revision surgery approximately 2 weeks later. The subject had various comorbidities including obesity (BMI = 36.6) and diabetes. No subsequent surgical interventions were associated with the expandable interbody fusion device.
DISCUSSION
Prior to the adoption of any novel technology, such as a biplanar expandable interbody device, a critical evaluation of its clinical and radiographic outcomes is important to establish an evidence-basis for safety and effectiveness. The PLIF and TLIF fusion rates cited by recent meta-analyses ranged from 93% to 98% when pooled across studies.18–20 The current study reports successful fusion in 96.6% of patients (95% CI: 88.3%–99.0%) and 97.4% of levels (95% CI: 91.0%–99.3%), supporting a similar level of fusion success with the bidirectional expandable device (without the use of BMP or harvest of iliac crest autograft). These results were also commensurate with previous studies of expandable devices, which reported 12-month fusion rates ranging from 92% to 96%.9,21–23 Furthermore, Khechen et al8 reported the proportions of patients reaching MCID for ODI, VAS leg, and VAS back at 6 months follow-up after TLIF with an expandable device to be 60%, 70%, and 67%, respectively, which were similar to the 58%, 71%, and 76% observed in the current study at a mean follow-up of 4.5 months. Similarly, Alimi et al24 reported that 64%, 52%, and 52% of patients treated with an expandable device reached the MCID for ODI, VAS leg, and VAS back by an average of 19.3 months follow-up, but the MCID used for VAS was 3 points in that study.
It is evident that some of the procedure-related AEs, such as failure of posterior supplemental fixation and nonunion, may be dependent on multiple factors, including patient-related risk factors such as smoking and long-term steroid use.25 The popularity of minimally invasive TLIF techniques, typically involving a unilateral approach to the disc space, may limit the amount of disc space preparation and/or bone graft delivery, thereby impeding fusion rates. In addition, some expandable spacers with complex expansion mechanisms limit graft chamber volume and the ability to backfill the device with bone graft post-expansion. Open spacer design facilitates the efficient delivery of bone graft materials through the graft chamber and into the disc space following expansion.26 The bidirectional expandable device used in the present study uses an open design, which allows for bone graft distribution through the device and into the interbody space following insertion and expansion. Many expandable devices have complex internal mechanisms not only occupying the graft chamber but also giving rise to new modes of biomechanical failure that are not present with static interbody fusion cages. For example, Kim et al27 and Stein et al28 each observed postoperative retropulsion of a stackable PEEK layer used for expansion in 1 type of device. In studies8,9,21,29 of other expandable PLIF and TLIF devices, no device-related complications were observed intraoperatively or postoperatively. Similarly, the current study revealed no device-related AEs. However, procedure-related AEs that are known risks associated with PLIF and TLIF surgery in general were observed. The incidence and types of AEs observed in the present study are consistent with the rates and types of complications previously reported.3
There are several limitations of the current study. It is a multicenter retrospective case series without a control group with limited clinical follow-up, which may expose the results to potential selection bias, observation bias, and/or hindsight bias. These common forms of bias are inherent to retrospective, uncontrolled studies, particularly when follow-up is limited. Hindsight bias could be introduced when investigators adjudicate the intensity of an AE and whether an AE was device- or procedure-related, because the outcome is already known. To minimize this potential bias, definitions for device and procedure relatedness as well as AE intensity (none, mild, moderate, or severe) were provided in the protocol before performing the chart review. To minimize the potential for selection bias, the study drew from a consecutive series of patients at each site, only excluding patients from analysis on the basis of predefined eligibility criteria that were consistent with the FDA-cleared product labeling. Other retrospective studies on expandable interbody devices have observed similar limitations in follow-up: Massie et al23 reported only 54% data availability at 90 days follow-up, and Yee et al30 reported only 58% of patients with 1-year radiographs in their retrospective review. Limited follow-up can expose a study to observation bias if the reasons for patients not returning are nonrandom. For the current study, multiple logistic regression of patient and surgical variables was performed to determine whether there were any systematic differences between patients with versus without 12-month radiographs for fusion assessment. There were no significant differences in demographic or operative characteristics between the analyzed subgroups and patients without follow-up data.
CONCLUSION
This study provides preliminary clinical evidence for the safety and effectiveness of a novel bidirectional expandable interbody fusion device. Whereas PLIF and TLIF are well-established as mainstay treatment options for patients with lumbar degenerative pathologies, continued innovation in expandable spacers that support minimally invasive surgery, safer implantation, and optimal patient outcomes will be relevant in delivering value-based spine care. With no randomized controlled trials and relatively few prospective single-arm cohort studies, there remains a paucity of high-level clinical evidence on expandable PLIF and TLIF devices.8,9,21–24,30 Therefore, further prospective and randomized clinical trials are warranted to substantiate the positive, preliminary findings reported in the present study.
Footnotes
Disclosures and COI: The last author is a salaried employee of the contract research organization that received funding from the sponsor to conduct this study. Funding to support data acquisition and analysis was provided by Integrity Implants, Inc (Palm Beach Gardens, FL).
- This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery. Copyright © 2020 ISASS
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Biplanar Expandable Cages for Transforaminal Lumbar Interbody Fusion Are Safe and Achieve Good 1-Year Clinical and Radiological Outcomes in an Asian Population
- Biplanar Expandable Cages for Transforaminal Lumbar Interbody Fusion Are Safe and Achieve Good 1-Year Clinical and Radiological Outcomes in an Asian Population