


Abstract
Over the past few decades, remarkable advancements in the understanding of the origin of low-back pain and lumbar spinal disorders have been achieved. Spinal fusion is generally considered the “gold standard” in the treatment of low-back pain; however, fusion is also associated with accelerated degeneration of adjacent levels. Spinal arthroplasty and dynamic stabilization technologies, as well as the continuous improvement in diagnosis and surgical interventions, have opened a new era of treatment options. Recent advancements in nonfusion technologies such as motion-preservation devices and posterior dynamic stabilization may change the gold standard. These devices are designed with the intent to provide stabilization and eliminate pain while preserving motion of the functional spinal unit. The adaption of nonfusion technologies by the surgical community and payers for the treatment of degenerative spinal conditions will depend on the long-term clinical outcome of controlled randomized clinical studies. Although the development of nonfusion technology has just started and the adoption is very slow, it may be considered a viable option for motion preservation in coming years. This review article provides technical and surgical views from the past and from the present, as well as a glance at the future endeavors and challenges in instrumentation development for lumbar spinal disorders.
© 2011 SAS - The International Society for the Advancement of Spine Surgery. Published by Elsevier Inc. All rights reserved.
Implantable spinal devices have had humble origins. Spinous process wires and Harrington rods represent the birth of metallic fixation of the spine. Through countless iterations of improved designs, current spinal fixation systems have revolutionized the surgeon's ability to stabilize the spine through both anterior and posterior approaches. Despite origins in fixation, the most rapidly expanding category of spinal devices is those concerned with motion preservation. Interestingly, many of these new motion-preserving designs, such as posterior dynamic devices, have borrowed heavily from their rigid stabilization cousins in their methods of implantation and anchoring.
With this most obvious of distinctions made, spinal devices can be organized into 2 main types: (1) devices to effect fusion and (2) nonfusion, motion-preserving devices. Fusion devices are those that either directly or indirectly enable bone healing across a previously mobile motion segment. Some are intended to contain bone graft or an osteogenic substance, such as interbody cages or so-called vertebral body replacements. Others function to provide a mode of immediate and rigid immobilization, such as pedicle screws and anterior instrumentation systems, to correct spinal deformity, enhance bony healing, decompress neural elements, and/or allow for recovery. Indications can span the full gamut of disorders of the human spine.
Nonfusion methods have been suggested as an alternative to fusion for a variety of spinal disorders, including scoliosis, spinal stenosis, and discogenic low-back pain. Their common goal is to treat the underlying pathology by replacing proinflammatory tissues, restoring spinal alignment, and decompressing neural elements while preserving functional movement rather than obliterating it. In doing so, it is hoped that many of the problems associated with fusion can be obviated, such as prolonged recuperation time, bone graft donor-site morbidity, pseudarthrosis, and adjacent segment degeneration (ASD). Because their success does not rely on the often-unreliable occurrence of bone healing, nonfusion techniques may also lead to quicker recovery periods. Among the most popular indications for motionsparing technology has been the treatment of degenerative disc disorders of the lumbar spine, which is the focus of this report.
Categorizing current lumbar motion-preserving devices
Motion-preserving devices can be categorized into 4 main types: (1) total disc replacement (TDR), (2) prosthetic nucleus replacement, (3) posterior dynamic devices, and (4) facet replacement. Defining these different types of devices is a worthwhile exercise.
TDRs necessitate nearly complete removal of the disc. The implant contains endplate portions, usually constructed of a metallic composite, that are fixed to the vertebral endplates. The articulating portion of the device is interposed between the endplates and can be composed of various materials.
Contrasting TDR, prosthetic nucleus replacements necessitate evacuation of only the nucleus pulposus through an annulotomy. Otherwise, the annulus remains largely intact. The implant is usually a deformable material that is inserted into the disc space with or without a fixation method.
Posterior dynamic devices come in many forms. Dynamic, or so-called soft, stabilization and semirigid are both typically pedicle screw based. The screws are connected by a flexible longitudinal member. Currently available devices vary from articulating ball joints to longitudinal Silastic rods. Although they may be considered a separate category, interspinous process devices may also be considered posterior dynamic stabilization devices. These devices consist of an implant that is interposed between the spinous processes. Some are held in place by geometry alone, whereas others rely on supplemental tethering fixation.
There is some conceptual overlap between facet replacement and posterior dynamic stabilization. For the purposes of this article, facet replacement will be defined as those devices that replace and simulate the function of either the entire facet joint or its articulating surfaces. Although they may be marketed as facet replacements, devices that include a continuous connecting member between vertebral levels should more appropriately be termed dynamic stabilization devices.
Total Disc Replacement
The past
Initial concepts for TDRs arose more than 30 years ago. Because the intervertebral disc is a complex anatomic and functional structure, an efficient and reliable artificial disc is expected to reproduce the motion as well as bear the loads.1 Kostuik2 proposed a disc design with metal endplates held apart by a spring. Lee and Goel3 have documented 25 years of work on a TDR that uses an elastomeric material between the endplates, which has only recently begun investigational clinical trials. In 1978 Steffee suggested the use of a balland-socket joint that was cemented into the vertebral bodies with a posteriorly located center of rotation. Steffee's AcroFlex disc (AcroMed Corp, Cleveland, OH), which had a closed envelope to contain wear debris, was quickly abandoned after only a handful of clinical implantations. A myriad of other design patents have been submitted over the past 20 years that have never seen clinical fruition.4
The present
At the time of this writing, there are 2 TDRs approved for use by the Food and Drug Administration (FDA): In-Motion/Charité III from DePuy Spine, Raynham, Massachusetts, and ProDisc from Synthes, West Chester, Pennsylvania (Fig. 1). Within a discussion of “the present” designs, however, the prerelease history of these devices is important to understand.

Lumbar disc replacement: a, InMotion; b, Charité III; and c, ProDisc II.
In the early 1980s, Schellnack and Büttner-Janz designed the initial Charité disc replacement.5–7 They relied on concepts derived from total hip and knee prostheses that, for the most part, used polyethylene-on-metal articulating surfaces. Like the current version, the device had a freely mobile polyethylene core (Ultra High Molecular Weight Polyethlene [UHMWPe]) that has convex surfaces, which articulate with concave metallic endplates (Charité I and II used stainless steel whereas Charité III and InMotion use cobaltchromium-molybdenum (CoCrMo) with titanium calcium phosphate coating). The small size of the Charité I implant led to a high frequency of subsidence. The Charité II attempted to remedy this problem by including thin lateral wings to increase the surface area. Unfortunately, these wings developed early fractures. The third version of Charité was developed in 1987. It featured a substantially broadened, flat endplate design that minimized endplate subsidence and fractures that were observed with the first and second generations. In 2007 the InMotion disc replacement was released to facilitate the insertion of the Charité III. The new InMotion design retains the essential characteristics of the Charité III design with only the modifications involving the teeth orientation and the addition of a central rail portion allowing the prosthesis to glide onto a ramp inserter.
The endplate of the InMotion and Charité III is manufactured from a CoCrMo alloy. This material has been found to result in less polyethylene wear debris than titanium or stainless steel in total hip and knee replacements. To maximize osseous integration, the endplates have a porous coating made of titanium and a layer of calcium phosphate. The design improvements of the current model appear to successfully address issues of implant subsidence and fatigue fracture. Analyses of revision cases have shown acceptable levels of polyethylene debris and substantial percentages of bony ingrowth.8, 9 The design of the core is intended to prevent core migration; reports of device migration could be due to excessive resection of the annular ring and positioning the device in hyperextension. Complications with the Charité III from Europe have much to do with (1) the implant endplate sizes offered at that time and (2) the lack of understanding of the best clinical indications and refined surgical techniques and instruments.10
In comparison to most other TDR designs, the distinguishing design feature of the Charité III is a sliding, freely mobile polyethylene core. With this design, the instantaneous axis of rotation is permitted to translate anterior and posterior to the midpoint of the disc during extension and flexion, respectively. Cunningham11 showed that the motion of the Charité III disc closely parallels normal motion in a cadaveric model. He found that the disc restored motion to that of the intact segment in flexion-extension and lateral bending; however, axial rotation exceeded normal limits. In addition, he found that the Charité III preserved the segmental kinematics and centroid of intervertebral motion at the operative and adjacent spinal levels compared with conventional methods of stabilization.
Early and medium-term clinical results with the Charité III for the treatment of low-back pain from degenerative disc disease (DDD) have been encouraging.12 Review of these results led to its approval as the first TDR for general use in the United States by the FDA, in 2004, as an alternative to lumbar fusion. Though not statistically different, results in the Charité patients tended to be better in this highly select group. Complication rates were similar in the TDR and fusion groups. Long-term results are available from Europe. With a minimum follow-up of 10 years, Lemaire et al13 and David14 reported good or excellent clinical outcomes in 90% of patients. Furthermore, they showed a mean of 10.3° of motion in flexion-extension at follow-up. Clinical failures of the Charité III have been reported recently.10, 15 Most of these failures could be linked to the application of the artificial disc in what is considered a contraindication in the FDA investigational device exemption (IDE) study.
The history of the ProDisc device has followed a similar path. In the late 1980s, Marnay developed the initial prototypes of the ProDisc. After the initial introduction of the ProDisc I, the ProDisc II was developed. In contrast to the Charité, the current ProDisc implant relies on a single articulating interface between the superior metallic endplate and the superior surface of the polyethylene core. It can be considered a semiconstrained device; the polyethylene core (bearing surface) is fixed to the inferior endplate but not to the superior endplate. Fixation of the endplates is enhanced by a single midline sagittal keel and 2 small spikes, in contrast to the 6 small teeth of the Charité III.
There are biomechanical differences between the InMotion/Charité III and the ProDisc II. By design, with the use of a fixed center of rotation, there is an obligatory amount of sagittal translation that is produced on the ProDisc during flexion and extension. This is a product of a rotational axis that lies within the anterosuperior aspect of the lower vertebral body. It is thought that this could produce abnormal forces within the facet joints and anteroposterior dimensional changes of the neuroforamina during sagittal motion.
Short- and long-term follow-up studies have shown good clinical results with the ProDisc II.16–18 Radiographic analysis has shown at least 2° of sagittal motion in 66% of prostheses after a mean of 8.7 years; the mean range of motion of all prostheses was 3.8°. The prevention of ASD has been one of the primary impetuses for TDR development, as ASD was reported in 24% of fusion cases, although no patients required surgical intervention. Because its prevention has been one of the primary impetuses for TDR development, ASD was reported in 24% of cases, although no patients required surgical intervention.19 As with the Charité III, complications have been reported. The size of the endplate keel has been reduced after reports of vertebral body–splitting fractures during insertion. The ProDisc was approved by the FDA in 2006.
There are a number of other lumbar TDRs that have been under investigation and are emerging onto the market, such as the Maverick (Medtronic Sofamor Danek, Memphis, Tennessee), FlexiCore (Stryker, Allendale, New Jersey), Kineflex (SpinalMotion, Mountain View, California), Mobidisc (LDR, Troyes, France), XL TDR (NuVasive, San Diego, California), Freedom Lumbar Disc (Axiomed Spine, Cleveland, Ohio), Activ-L (Aesculap B. Braun, Tuttlingen, Germany), and Triumph (Globus Medical, Audubon, Pennsylvania) (Fig. 2). This device list is unlikely exhaustive but aims to offer an overview of the current technologies available on the market today. Although they all have some unique features, their general design concept is that of a semiconstrained TDR with articulating surfaces. Clinical results appear to be similar to those for the Charité and ProDisc20; however, a thorough comparison between implant designs and associated clinical outcomes has yet to be published.

Lumbar disc replacement: a, Maverick; b, FlexiCore; c, Kineflex; d, Mobidisc; e, XL TDR; f, Freedom; g, Activ-L; and h, Triumph.
The Maverick consists of 2 metallic CoCrMo endplates that directly create a ball-and-socket system with the rationale to minimize wear, a posterior center of rotation to match that of the disc segment aimed at unloading the facet joint, and hydroxyapatite (HA) coating on the bone-implant interface to maximize early fixation with bony ingrowth. CE mark was obtained in 2001, and the prosthesis has been in clinical use since 2002. The first FDA clinical study started in 2003 and closed in 2010 with positive results. FDA approval is still pending at this stage, and a second US study started in 2009 with estimated study completion in 2013.
The FlexiCore disc is a 2-piece constrained metal-onmetal device designed as a ball-and-socket joint where no translation of the endplate is allowed. The clinical IDE enrollment is completed. Three-year preliminary results are being presented as encouraging, but full study data are still required to confirm these.
The Kineflex is a 3-piece metal-on-metal design (2 keeled CoCrMo endplates and 1 semiconstrained, fully articulating CoCrMo core) that aims to provide a freedom of movement with translational stop. It has been used since 2002 outside the United States. An FDA IDE study commenced in 2005 with a comparison to the Charité TDR and closed in 2006, with follow-up on the over 500 patients up to 3 years at this stage. FDA approval has not been granted yet.
The Mobidisc is a 3-piece design with 2 metallic endplates with a modular keel and a polyethylene core with geometries allowing an on-constraint prosthesis with controlled mobility in all axes. The device was released in 2003. It is currently not available in the United States.
Other devices, such as the XL TDR, have a metallic design that is implanted from the patient's side, with a similar approach to Extreme Lateral Interbody Fusion (XLIF) cages. An FDA trial started in 2009 with an estimated primary completion date in 2012.
The Freedom Lumbar Disc is made of a silicone polycarbonate-urethane polymer core that is bonded between the 2 metal endplates to allow controlled motion and enable shock-absorption capability. The device has been used in Europe, and FDA trials started in 2008 with an estimated primary completion date in 2012.
The Activ-L prosthesis consists of a polyethylene core that has controlled anterior-posterior translation capability between 2 metallic endplates that can be inserted from an anterior, anterior-lateral, or lateral approach. An FDA trial started in 2007, enrollment was completed and currently it is in the final stages of data collection and submission.
The Triumph consists of 2 metallic endplates with a geometry allowing it to be inserted through a posterolateral approach. An IDE study started in 2007; it is still ongoing but no longer recruiting.
At the time of this writing, there are also other devices either under clinical investigation outside the United States or available internationally, such as the Dynardi Artificial Lumbar Disc (Zimmer, Warsaw, Indiana) with 2 metallic endplates on a polyethylene-core design, Lumbar Motion Preservation (LMP; Vertebron, Stratford, Connecticut), CadiscL (Ranier Technology, Cambridge, United Kingdom) with a polyurethane-polycarbonate graduated modulus monobloc design, Physio-L (Nexgen Spine, Whippany, New Jersey) with a titanium porous endplate directly linked onto a polycarbonate-polyurethane core, Total Spinal Motion Segment System (TSMS; Disc Motion Technologies, Boca Raton, Florida) with a metallic rod with a built-in damper, eDisc (Theken Disc, Akron, Ohio) with a polymer bounded between metallic endplates with additional microelectronics allowing activity monitoring, Welldisc (Eden Spine, Lake Mary, Florida) with a metallic endplate and polyethylene core, La Jolla (Seaspine, Vista, California), and M6L (Spinal Kinetics, Sunnyvale, California) with a shock-absorbing core encapsulated within a polymer sheath mimicking the annulus fibrosus bound into metallic endplates.
The future
Notwithstanding material and specific design differences, the motion-producing mechanism of most TDRs is essentially a ball joint. Though much more sophisticated, this is quite analogous to Fernström metal balls. Other features such as elastic shock-absorption properties are not approximated with any of these designs. LeHuec et al21 have shown that there is no difference in the shock absorption provided by a hard polymer core or a metal-to-metal device. However, this will likely be a focus of future designs. There are a number of ways that axial compressibility (shock absorption) can be achieved in a TDR. In his early patents, Kostuik2 described the use of a spring. This is not likely to be adopted, because there are a finite number of cycles before which a metallic spring will fail. Other investigators have proposed the use of a rubbery-type substance.
Limitations in material design led to failure of the AcroFlex disc in the 1990s. Most recently, Lee and Goel3 have finally introduced their TDR design that uses an elastomeric core material that is affixed to the metallic implants by a proprietary process. Interestingly, such a design has inherent limits to range of motion; it has been proposed that the material and structural properties of the elastomeric material have been optimized to closely approximate those of the intact disc. However, creep deformations and hysteresis properties of elastomeric materials may be limiting factors in this application.
Future advancement of lumbar TDR may have little to do with the design of the device. Because difficult revision surgery may be a factor limiting general acceptance of TDR, methods of scar prevention between the spine and the neighboring retroperitoneal structures have been sought. This may be in the form of biologically inert or active membranes.
Nucleus Replacement
The past
In considering Fernström balls, one might be hard-pressed to describe them as a TDR according to the definitions described previously. More accurately, they may be described as a nucleus replacement. As such, Fernström balls are the predecessors of modern nucleus replacements. A subset of nucleus replacement is nucleus augmentation, which may be defined as those substances that are injected into the disc space and cured in situ. This seemingly novel idea has its roots in Nachemson's attempt to augment a degenerated disc with injected silicone.
The present
Although there are no nucleus replacements or augmentation devices that are currently approved for use in the United States, there are a number that are under clinical investigation.
Ray22 designed the original Prosthetic Disc Nucleus (PDN; Raymedica, Minneapolis, Minnesota) as well as several subsequent modifications (Fig. 3). The PDN is intended to be a treatment for discogenic low-back pain from DDD or after discectomy. In distinction to TDRs, the PDN is placed through a posterior laminectomy and standard discectomy/ annulotomy approach. The PDN's essential component is a hydrophilic gel pillow that absorbs water and expands once implanted. The gel pillow is contained by a polyethylene mesh “pillow cover” to prevent overexpansion. The initial design that was clinically released in 1996 included 2 pillows placed transversely within the disc space. Current versions call for right and left pillows to be placed in an anteroposterior orientation.

Nucleus pulposus supports: a, PDN; and b, DASCOR.
One of the major design challenges of any nucleus replacement or augmentation method is to prevent expulsion of the device from the disc space. Migration and subsidence with the first-generation PDN were reported. Thus subsequent versions, such as PDN-SOLO and PDN-SOLO XL, include a suture fixation method. In addition, the insertion approach has been changed. Using a direct lateral approach avoids an open annulotomy within the epidural space. Early clinical results have been encouraging. Pain relief has been reported with the PDN at 6 months and 1 year of followup.23, 24 However, migrations have continued to occur25 with disc herniations. Raymedica has reported that over 4,000 patients worldwide have received either the PDN or a PDN-SOLO implant. Only recently has the PDN-SOLO device been implanted in the United States as part of a nonrandomized, prospective clinical trial. In 2006 Raymedica released the Hydraflex system, with a more anatomically profiled implant, softer core, larger footprint, quicker absorption capability, and specially dedicated instrumentation to facilitate repeatable meticulous implantation aimed at reducing the extrusion complication rate.
The DASCOR Disc Arthroplasty System (Disc Dynamics, Eden Prairie, Minnesota), which is another PDN device, uses an in situ, curable polymer and is indicated for treating patients with chronic low-back pain caused by DDD. This system has been CE marked in Europe, and the clinical investigation in the United States stopped enrollment in early 2007. The DASCOR device consists of a 2-part curable polyurethane and an expandable polyurethane balloon, which is inserted into the disc nucleus space after the nucleus has been removed.26 The balloon is then injected with the flowable polymer, which creates a complete patient-specific implant that conforms to the shape and size of the disc space. The flowable polymer cures, creating a firm but pliable implant. Disc Dynamics reported that clinical study results using the DASCOR Disc Arthroplasty System have shown that most patients have had improvement in pain and function.27 Because no 2 patients recover at the same rate, no guarantees can be given on the rate or degree of pain relief each individual can expect. Disc Dynamics started to liquidate its assets in 2009, and currently, support on the IDE study sought for its DASCOR device remains unclear.
The NeuDisc spinal nucleus implant (Replication Medical, Cranbury, New Jersey) is designed to replace the native nucleus pulposus, restore function to the disc, and slow or reverse the degenerative process. The NeuDisc is intended to be implanted through standard minimally invasive surgical procedures. The device is a small, coin-sized implant composed of 2 grades of a modified hydrolyzed poly-acrylonitrile polymer (Aquacryl, Replication Medical, Cranbury, New Jersey). The Aquacryl polymer is reinforced by a Dacron mesh. It is inserted in a dehydrated state, which allows the implant to be placed via minimally invasive methods. Once inserted, it absorbs up to 90% of its weight in water in an anisotropic fashion to restore disc height and improve compressive axial load resistance.28 This hydrogel material device has the advantage of allowing fluid communication and potential nutrient flow between the vertebral bodies, similar to the native nucleus.
The NUBAC disc arthroplasty system (Invibio, Green-ville, North Carolina/Pioneer, Driebergen, The Netherlands) has been designed to restore and maintain load-sharing capabilities of the implanted disc. It is a polyether ether ketone (PEEK)–on–PEEK system with articulating surfaces and an endplate that is slightly concave. PEEK has an elastic modulus similar to underlying bone and magnetic resonance imaging compatibility. Implantation is less invasive with this nucleotomy procedure. The relatively small bearing implant system is placed in the center of the endplate and is expected to resist expulsion well. A clinical trial study was initiated in the United States in 2009, and the most recent update, from 2010, indicated that the study was still ongoing but no longer recruiting patients.
The NuCore Injectable Nucleus (Spine Wave, Shelton, Connecticut) is designed to provide patients and surgeons with a minimally invasive surgical alternative for treating the early stages of DDD and as an adjunct to microdiscectomy (Fig. 4). The NuCore Injectable Nucleus is an injectable, 100% synthetic recombinant protein hydrogel designed to mimic the natural nucleus and restore the spine's natural biomechanics. This device is designed with the premise to restore the biomechanics of the disc after micro-discectomy and reduce accelerated disc degeneration. Bio-mechanically, the hypothesis regarding NuCore is that it will augment the remaining nucleus material and restore the spine's natural biomechanics.

Nucleus pulposus alternatives: a, NuCore; b, NeuDisc; c, NUBAC; and d, PDN.
TranS1 (Wilmington, North Carolina) has released the Percutaneous Nucleus Replacement (PNR). It consists of a titanium screw system anchoring itself onto the superior and inferior vertebrae, with a central membrane that is filled with curable material and acts as the nucleus. It is inserted in a caudal-rostral manner anterior to the sacrum axially through the vertebra to replace the damaged disc and restore the natural motion while also preserving the integrity of the annulus fibrosus and ligaments so as to reduce the risk of implant migration. Because of the approach, the system is limited to L5-S1. It has been in limited use since 2008, and its clinical performance has yet to be evaluated.
At the time of this writing, there are also other systems available such the BioDisc (CryoLife, Atlanta, Georgia), DiscCell (Gentis, Wayne, Pennsylvania), and GelStix (Replication Medical).
The complications of nucleus replacement devices typically includes: 1) implant expulsion, especially with those devices without retaining means, because the annulus fibrosus is weakened to allow insertion; 2) subsidence, especially in stiff devices because of high stress created in the center of the endplate; 3) endplate remodeling with loss of disc height; and 4) poor disc kinematics resulting in neural impingement and facet degeneration or fracture.
The future
Assuming nucleus replacement and augmentation devices will be approved for use in the United States, a future challenge will be to distinguish which patients are ideal candidates. At this time, it is unclear who would benefit from a TDR versus a nucleus replacement.22 Some investigators have considered advanced disc space collapse (<5 mm of residual disc height), endplate defects, and obesity (body mass index >30) to be contraindications to nucleus replacement. Despite these mechanical considerations, more fundamental questions regarding low-back pain generators remain unanswered.
New generations of injectable polymeric nucleus devices that can cure in situ are in the beginning phases of development. Hydrogel materials intended to restore the disc height by supplementing the dehydrated and degenerated nucleus pulposus are also in development. The goals of these devices are to restore physiologic intervertebral ligamentous stress and annular tension to re-establish spinal kinematics. One of the impetuses for developing injectable devices is to lower the chance for extrusion, because they are implanted through a smaller needle-stick annulotomy. In addition, insertion is potentially less invasive than with nucleus replacement. Other challenges arise, though, such as the ability to maintain disc space height long-term, achieving adhesion to the host tissue, and creep. As a minimally invasive procedure, its future role may be to delay or mitigate the need for open surgical treatment of discogenic low-back pain.
There are a number of devices and drugs that are currently in development. Intradiscal rhGDF-5 is a new drug being developed for the treatment of early DDD. The active pharmaceutical ingredient is recombinant human growth and differentiation factor 5 (rhGDF-5). Growth and differentiation factor 5 is a member of the transforming growth factor β superfamily and the bone morphogenetic protein subfamily and is known to influence the growth and differentiation of various tissues. Preclinical studies suggest that intradiscal rhGDF-5 is safe in the intended route of administration. Moreover, preclinical studies in animal models of disc degeneration have shown the ability of rhGDF-5 to reverse the degenerative changes as evidenced by an increase in disc height and hydration, as indicated by T2-weighted magnetic resonance imaging, as well as reduce the cytokine expression in degenerative animal discs. Currently, rhGDF-5 pilot clinical studies are being conducted to prove the safety and efficacy before pivotal studies.
Another area of future development will be techniques for annular repair or reconstruction. Sealing the annulus after nucleus replacement might have benefits in diminishing the chance for extrusion. The key types of surgical sealants are fibrin sealants, synthetic agents, collagen-based compounds, and tissue adhesive glues such as hydrogel. Fibrin sealants are popular because of their safety, low risk for infection, and promotion of natural tissue healing. Such barriers have shown decreased scar formation in animal models.29
Furthermore, annular reconstruction may an attractive method of decreasing the rate of reherniation after standard discectomy. However, the annular ring is subjected to pressures as high as 3.5 MPa, so annular repair must be robust enough to withstand these high pressures and prevent disc reherniation. Other theoretic benefits have also been proposed, such as containment of water and neural inflammatory factors within the disc, which might reverse or arrest disc degeneration and lessen radiculopathy, respectively.
Posterior dynamic stabilization devices
Posterior nonfusion stabilization of the lumbar spine had been infrequently practiced before the last decade.30, 31 Early attempts ranged from tethering artificial ligaments to interspinous distraction devices. Early failures prevented further interest until recently, although posterior dynamic stabilization has become one of the most rapidly evolving fields in spinal surgery. This new category of thoracolumbar spinal surgery focuses on the concept of maintaining or restoring intervertebral motion in a controlled fashion, whether by restricting spinal motion or by dampening the kinetic energy involved in motion. The goal of these implantable devices is to restore the behavior of the spinal column and potentially prevent ASD.32
With greater focus on motion-preservation alternatives, interest in posterior dynamic stabilization has grown. Various devices have sought a wide array of indications, including accelerated fusion, treatment of discogenic low-back pain, treatment of facet pain, treatment of spinal stenosis, controlling motion in degenerative spondylolisthesis or iatrogenic destabilized spine, treatment of neurogenic claudication, and prevention of ASD at the time of fusion.
Interspinous devices
The past: What is old is new; more importantly, what is new is old. Although the surge of interspinous devices to the spine market would have the casual observer convinced that they are the product of a recent brainstorm, it is not the case. The notion of an interspinous device to produce segmental posterior distraction was first introduced in the 1960s by Dr Fred Knowles. Better known for his hip pin design, he reported limited success with his spinal device because of subsidence and displacement. He would not have seen the reincarnation of his ideas in the form of an X-stop (Kyphon, Sunnyvale, California) for 3 decades.
The present: Several types of interspinous devices have been developed. At the time of this writing, X-stop is the only approved device. Others, such as the Wallis system (Abbott Spine, Austin, TX; acquired in 2008 by Zimmer), Minns silicone distraction device, CoFlex system (Paradigm Spine LLC, New York, New York), and DIAM (Medtronic Sofamor Danek) are under clinical investigation with the intent to seek FDA approval (Fig. 5). They all work to limit spinal extension. These interspinous spacers may be helpful when more conservative (nonoperative) care does not improve symptoms. They can be used in elderly adults who are not well enough to undergo surgery; indeed, most recent devices can be inserted without tissue or bone removal and without the need for general anesthesia. The indications for these devices remain to be defined. Manufacturers’ reported indications include DDD, ASD, facet syndrome, spinal stenosis, and nontraumatic instability. Interestingly, some devices, such as the CoFlex, have sought approval as fusion devices because they are constructed of allograft bone.

Interspinous spacers: a, Wallis system; b, DIAM system; c, X-stop; and d, CoFlex.
All these devices hold the spine in a position of slight flexion to decompress the spinal cord or spinal nerve roots. The spine can still rotate or bend to the side when the spacer is in place. All interspinous spacers can be categorized into 2 groups: static spacers allow constant extension, whereas dynamic spacers are compressible by use of an elastomeric material.
The X-stop is made of titanium and PEEK components, with side wings encapsulating the lateral sides of the spinous processes to reduce the risk of implant migration. FDA approval was obtained in 2005 after a 2-year clinical study. The device is approved for use in patients aged 50 years or older with lower-extremity neurogenic pain from lumbar spinal stenosis and can be inserted under local anesthesia. The ligaments around the spine are saved from being cut. The device slips through a slit made in the ligament. In the pilot study, inclusion criteria were mild or moderate symptoms that were relieved by flexion and the ability to walk at least 50 ft. Exclusion criteria were a fixed motor deficit or spondylolisthesis of grade II or higher.33, 34 Clinical studies by Anderson et al35 showed overall clinical success in 63.4% of the study group but in only 12.9% of the control group at 2-year follow-up. Clinical failure includes laminectomy and fusion with no evidence of any nerve injuries or neurologic deterioration. At present, 30,000 patients have been treated with X-stop.
The Wallis system was first released clinically in Europe in 1987 under the form of a titanium spacer design that has since been replaced by a plastic-like polymer material (PEEK) that is secured between the spinous processes with a woven polymer flat band so as to reduce implant migration risk and constraint motion to prevent extreme movements. FDA clinical trials started in 2007 and are currently at the stage of final data collection for primary outcome measurement.
The DIAM system is made of a silicon H-shaped spacer encased within a polyester jacket that is secured (after removal of the interspinous ligament) with a cord similar to the Wallis system's. The first clinical case was performed in 1997 in France, and 25,000 patients have been treated outside the United States since then. FDA clinical trials have also started, such as the study relating to the effectiveness of DIAM versus decompression versus posterolateral fusion that started in 2008 and closed in December 2010 with final data collection for primary outcome measurement or the study relating to the effectiveness of DIAM versus conservative care that started in 2007 and has a primary completion date estimated for 2013.
The CoFlex is based on the interspinous-U design from Fixano (Péronnas, France) that was clinically used from 1995 onward. It is made in its classic form as a titanium U-shaped metal design that is maintained between spinous processes with side wings, so as to control movement while allowing motion, being marketed as a nonfusion device. The CoFlex-F device has also been released. On the basis of the CoFlex design, bolts are added through the holes of the wings to rigidly secure those onto the spinous processes; it is indicated for fusion, as an alternative to pedicle screw fixation as an adjunct to interbody fusion. The IDE study started in 2007 and is still recruiting patients.
The future: There are many interspinous distraction devices planned for future study. Some are compressible, such as the CoFlex, DIAM, or Minns devices,36 whereas others are noncompressible. Fixation techniques also vary. Although the rationale for their use in the treatment of spinal stenosis is clear, the role in the treatment of DDD remains to be defined. One proposed mechanism of action is unloading of the posterior annulus by distraction. Interspinous devices with shock absorption and postoperative adjustability may present the future of these devices.
In recent years multiple companies have offered various devices, such as NuVasive with ExtendSure; Biomech's (Taipei, Taiwan) Promise and Rocker designs, made of PEEK and mobile core and articulated design, respectively; Cousin Biotech (Wervicq-Sud, France) with Biolig silicon encapsulated in woven synthetics; Alphatec (Carlsbad, California) with the HeliFix screw-type PEEK space design; Vertiflex (San Clemente, California) with the Superion implant whose deployable wings aim at less invasive insertion (IDE study started 2008); Synthes with the In-Space system with minimal insertion (IDE study also started in 2008); Medtronic's Aperius PercLID implant (IDE study started in 2009), Orthofix (Bussolengo, Italy) with InSWing; Pioneer with BacJac; Maxx Spine (Bad Schwalbach, Germany) with I-MAXX; Sintea Plustek (Assago, Italy) with Viking; Globus Medical with Flexus; and Privelop (Neunkirchen-Seelscheid, Germany) with Spinos (Fig. 6).

Other interspinous spacer alternatives: a, Promise; b, Rocker; c, Biolig; d, HeliFix; e, Superion; f, In-Space; g, Aperius; h, InSWing; i, BacJac; j, I-MAXX; k, Viking; l, Flexus; m, Spinos; and n, Wellex (Eden Spine).
The typical complications of interspinous devices include spinous process fracture, especially with stiff design; novel radiculopathy, especially with devices with limited motion-constraining ability; and returning or increased pain around the implant area. Implant dislodgement is also a potential complication, particularly in those designs with limited fixation means.
Posterior dynamic, or “soft,” stabilization
The past: The Graf ligament is one of the earliest attempts at posterior dynamic stabilization (PDS) (Fig. 7). Developed in France by Dr H. Graf, it included artificial ligaments that were fixed to pedicle screw anchors. The ligaments were tensioned to provide stability. Clinical reports of its use have been published with mixed results.37–39 Some of the variation may be from variability of indications. However, device-related complications are not uncommon. Over-tensioning of the ligament has led to foraminal stenosis and new-onset radiculopathy. Pedicle screw loosening has also been reported.
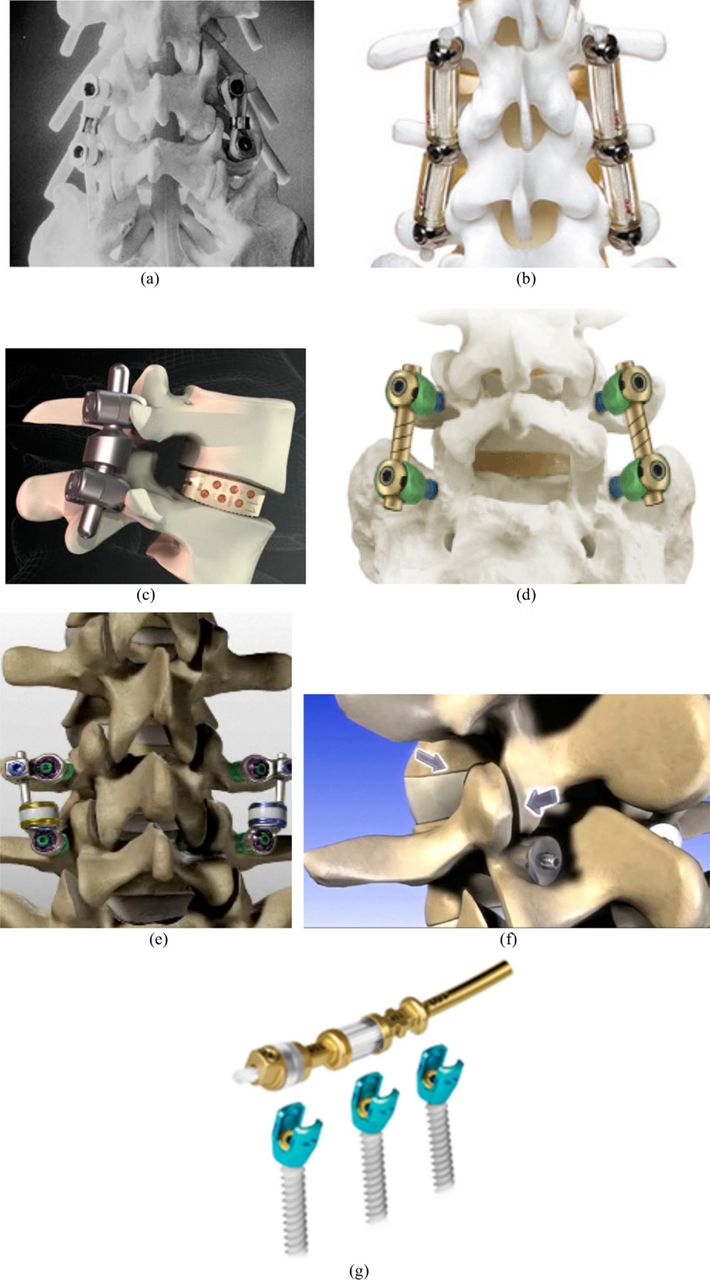
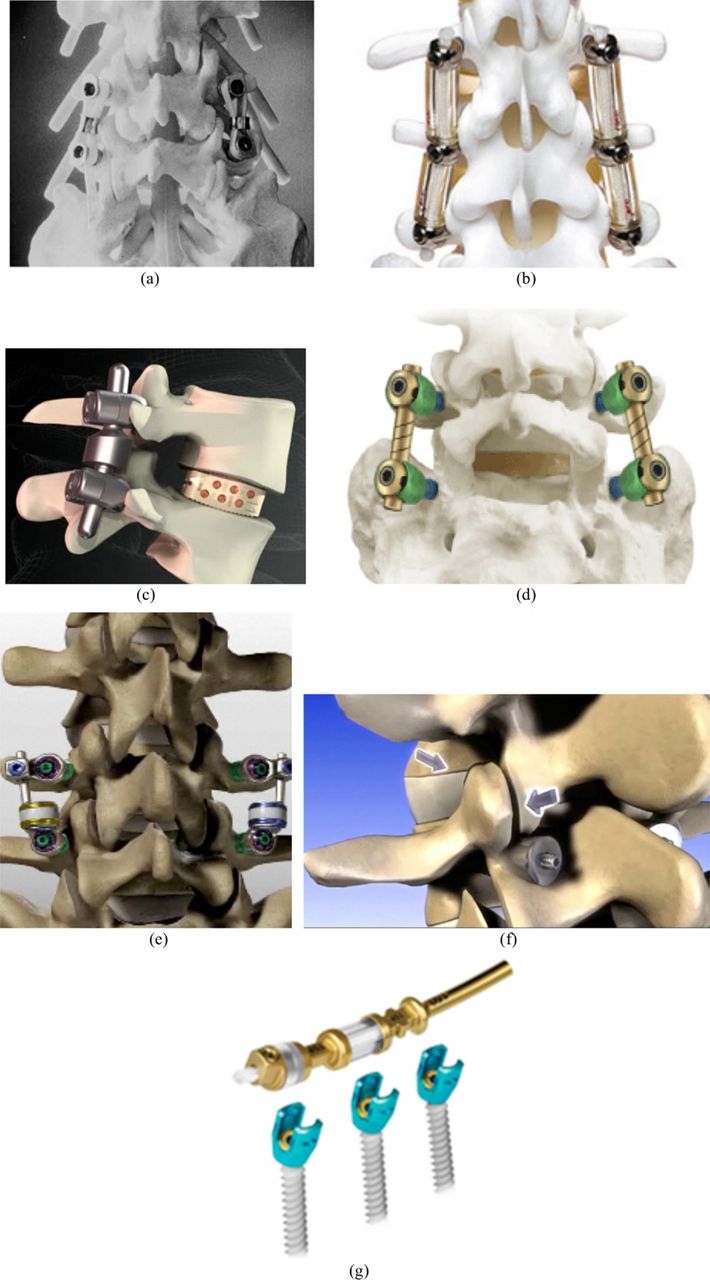
Posterior dynamic stabilization: a, Graf system; b, Dynesys; c, IsoBar; d, AccuFlex; e, Stabilimax; f, Percudyn; and g, Transition.
The present: Current PDS systems purport the ability to allow controlled motion in such a way as to achieve more normal movement of the spine. The Dynesys system (Zimmer Spine, Warsaw, Indiana) was developed by Gilles Dubois in 1994. In its present form, the system consists of titanium alloy (Protasul 100) pedicle screws and a stabilizing spacer made of polyester (Sulene-PET) cords and a polycarbonate-urethane (Sulene-PCU) tube. The spacer fits between and is fixed to the pedicle screw heads. In extension, the tube resists compression; in flexion, the polyester cord resists tension. The device has been used in Europe for a variety of indications with mixed results.40, 41
The IsoBar (Scient'X, Beaurains, France) and AccuFlex (Globus Medical) rod systems are other examples of PDS systems. These devices’ rods rely on a flexible section of rod to provide a low stiffness connection between screws. They provide less rigidity, permitting load sharing with the anterior column. Both systems received FDA clearance as fusion devices. The IsoBar relies on a damper system (stacked titanium disc-shaped elements) that allow micromotion of the rod; it has been used since 1993 in Europe and became available in 2003 in the United States, although the FDA has issued warning letters in 2010 and 2011 to question the postmarket surveillance plan in place. The manufacturer is currently indicating that the product promotes stimulated and improved fusion. The AccuFlex semirigid system is created by a helical cut within a standard metallic rod. There has been concern about the issue of ingrowth within the spiral that might prevent proper functioning of the device throughout time. Globus Medical currently also offers the Transition system claiming to optimize load sharing while ensuring transitional stabilization, with a design appearing to be similar to that of Dynesys. In 2010 it was announced that 2,000 patients had already been treated with this device. To date, however, there are no peer-reviewed clinical articles on the use of any of the previously listed devices as nonfusion dynamic stabilization systems.
The Stabilimax (formerly known as the M-Brace; Applied Spine Technologies, New Haven, Connecticut), invented by Manohar Panjabi, has been specifically designed as a treatment for DDD and chronic low-back pain. This implant is unique in that relies on dynamization at the screw-rod interface as well as within the rod itself, a feature intended to lower the rate of screw loosening. Biomechanical studies in cadavers have shown that the device restores motion to the neutral zone even after bilateral medial facetectomy. As with most systems, it is pedicle screw based. In vitro studies have also shown minimal effects of the adjacent segments. Clinical investigation of this device is under way. An FDA study was started in 2007 aiming at terminating at the end of 2010, but it has been terminated because the company has ceased operation. Results were presented at the Spine Arthroplasty Society (SAS) conference in 2010. With 12 to 24 months’ follow-up on 23 patients, there was significant improvement postoperatively in comparison to preoperatively and there were 4 occurrences of device/ screw failures.
There have been efforts such as the Percudyn system (Interventional Spine, Irvine, California) consisting of a pedicle anchor upon which sits a polycarbonate-urethane stabilizer that serves as a mechanical stop between the inferior and superior facets to stiffen the facet column. Its bilateral fixation helps in stabilizing the spine in extension and lateral bending, and the bumper helps in offloading the facet joints by dissipating energy forces dorsally, reducing intradiscal pressure concomitantly. It is undergoing clinical studies outside the United States, and a 2-year follow-up study of 40 patients presented at the SAS conference in 2009 shows satisfactory improvement in pain.
In recent years the established companies have also launched PEEK rod systems sometimes accompanied by dynamic screws, such as the CD Horizon Legacy Peek Rod System (Medtronic, Memphis, Tennessee) and Expedium PEEK rod and Viper SC screw (DePuy Spine). Clinical outcomes have yet to be reported to advocate these systems for nonfusion treatments or topping-off long spinal fusion constructs.
The future: The major challenges of current and future PDS systems are to prevent implant and bone-implant interface failures and to show efficacy. With modern metallurgy techniques and bioengineering programs, devices will likely be able to closely approximate, if not match, normal physiologic motion and mechanics. However, this ability will be short-lived if mechanical failure is inevitable. This can occur between the bone and the implant or within the implant itself. More importantly, PDS systems must show efficacy for the treatment of lumbar spine disorders. If they are used as a post-laminectomy method of reconstruction, long-term clinical benefit by preventing instability and ASD must be shown. If they are to be used as a treatment for discogenic low-back pain, they must show equivalent outcomes to interbody fusion and/or TDR.
Facet replacement systems
The intervertebral disc is always considered as a source of back pain. As degeneration of the disc advances, facet load increases significantly,42 which ultimately leads to facet arthrosis, which may in turn cause low-back pain. Facet arthrosis is also a major contradiction for anterior disc replacement.43 Facet replacements can permanently remove the pain-generating articulation and capsule of the facet joints. To address facet pain, stenosis, and disc degeneration simultaneously, facet arthroplasty along with disc replacement may be a viable treatment option. Such a combination may restore motion and relieve patients from anterior, posterior, and neurogenic pain.
The past
Facet replacement patents appear as far back as the mid 1980s. However, it has not been until recently that devices under this category have been brought to market. Early proposed designs were primarily articular surface replacements, similar to those for peripheral joint replacements. Over time, designs have become larger and bulkier, replacing not only the articular surfaces but the entire joint and its supporting structures.
The present
The current role of facet replacement is multifaceted. It has been proposed as an adjunct to TDR, as a reconstruction method after laminectomy, and as a treatment for facetmediated pain.
One frequent contraindication for TDR is the presence of facet arthritis.43 Continued motion across arthritic facet joints is thought to be one of the causes of failed TDR surgery. A reliable method of replacing the facet joint would potentially avert this problem.
The so-called gold-standard reconstruction method after a destabilizing laminectomy is lumbar fusion, which is usually augmented with pedicle screws. Although this is a successful procedure with excellent clinical outcomes, there are a number of potential long-term drawbacks. ASD often occurs above the fused segment, which can eventuate in additional surgery.44 In addition, the immobilizing effects of fusion do not restore the normal function and mechanics of the decompressed segment, the ramifications of which are not well understood. Facet replacement has been proposed as an alternative to fusion and instrumentation after laminectomy for spinal stenosis. Trials are currently under way with a number of devices.
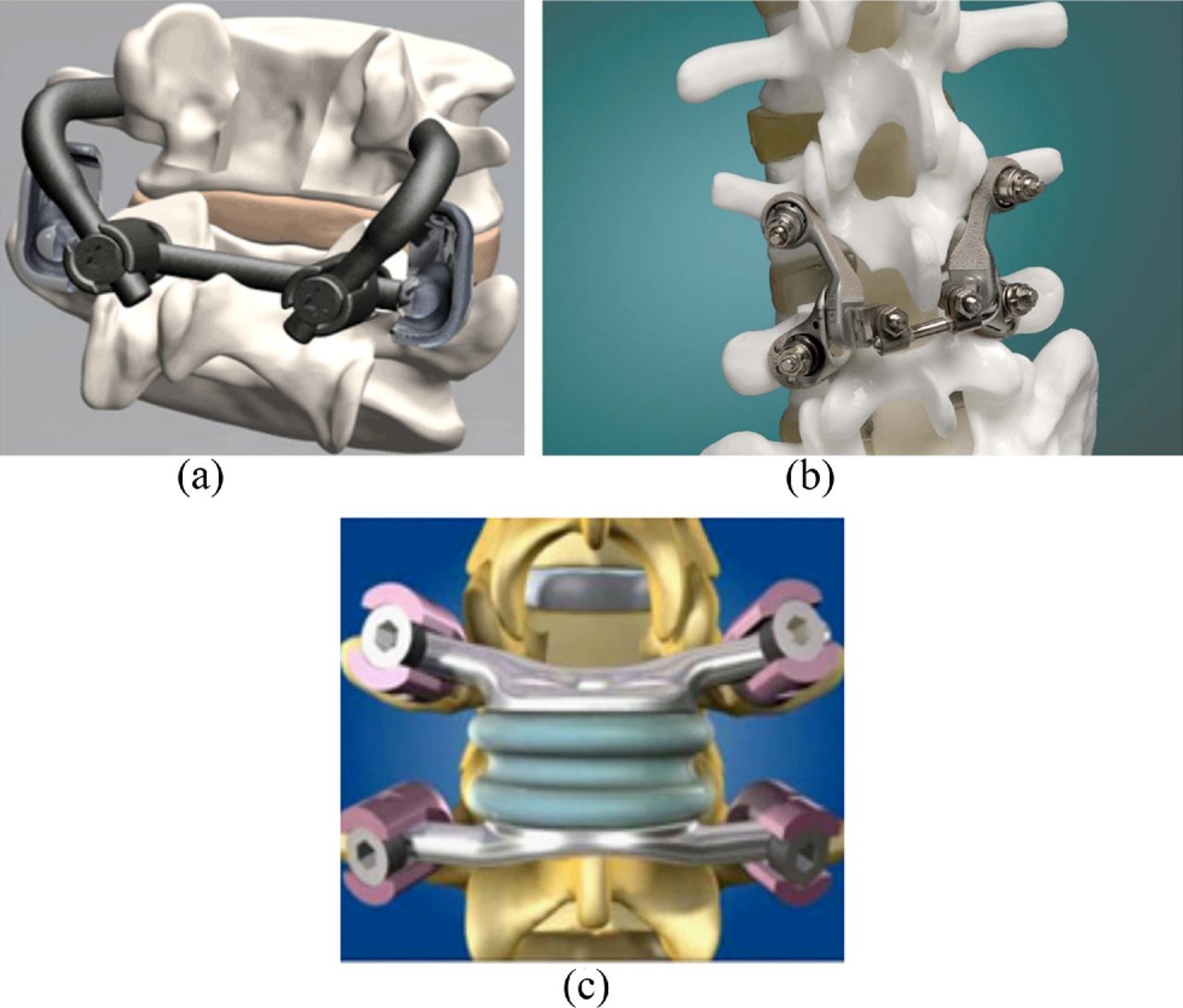
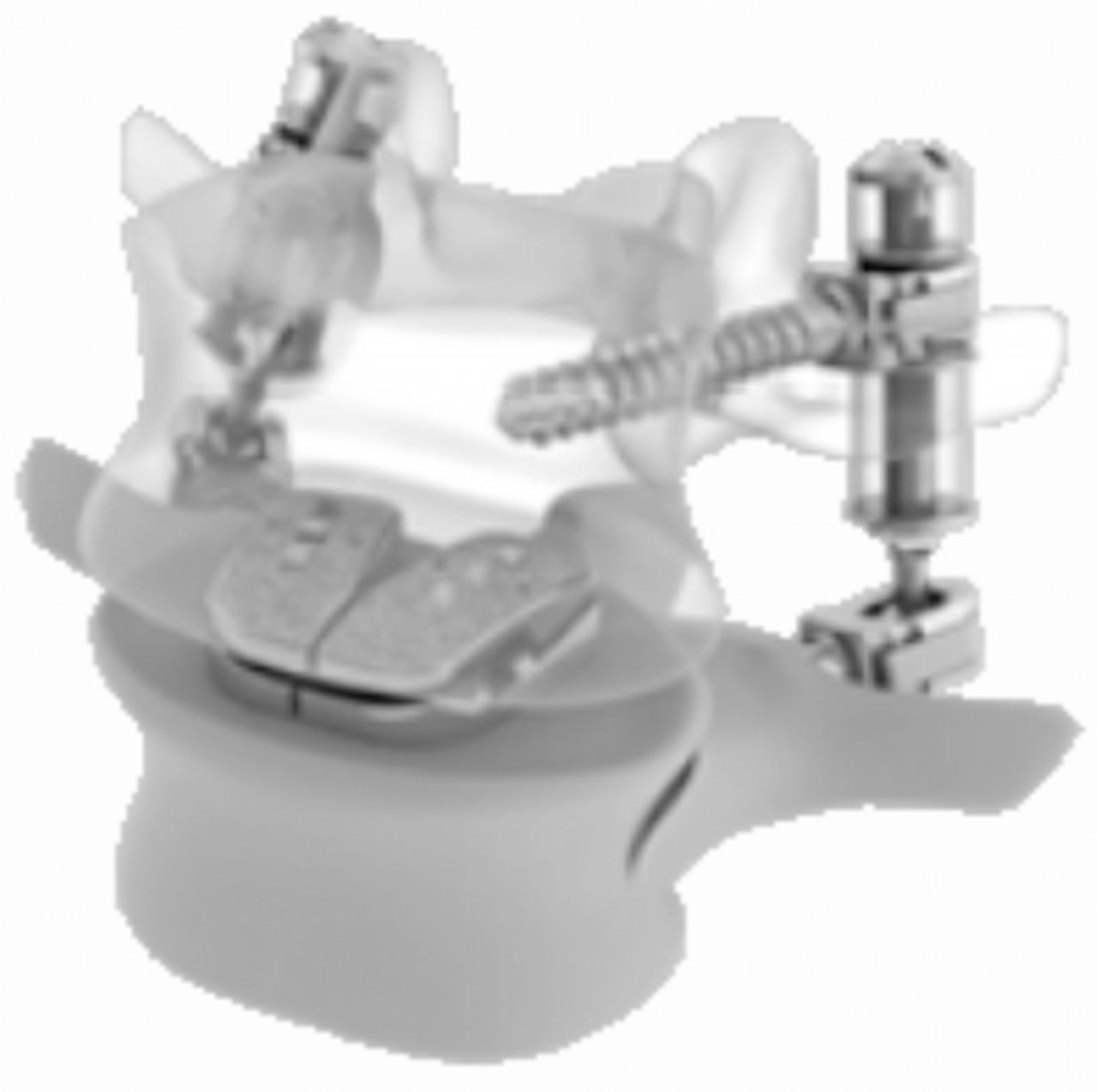
The facet replacement market segment is just now taking shape. So far, Archus Orthopedics (Redmond, Washington) has been a leader in the facet replacement market. One of their devices, called the Total Facet Arthroplasty System (TFAS), is a pedicle anchor– based system that relies on a sliding ball-in-bowl–type joint (Fig. 8). It is a nonfusion spinal implant indicated for treatment of patients with moderate to severe spinal stenosis. TFAS replaces the diseased facets (and lamina, if necessary to attain adequate decompression) after surgical removal. TFAS offers an alternative to rigid spinal fusion fixation enabling intervertebral motion and restoration of stability and sagittal balance to the spine. An IDE study was started in 2005 in which randomized patients with lumbar stenosis were treated with either TFAS stabilization or fusion with pedicle screws. The manufacturer underwent dissolution in 2009 and was acquired the same year by Facet Solutions (Hopkinton, Massachusetts), which was acquired by Globus Medical in early 2011. The current status of the IDE recruitment status is unknown as indicated on the National Institutes of Health FDA Clinical Trials Web site.
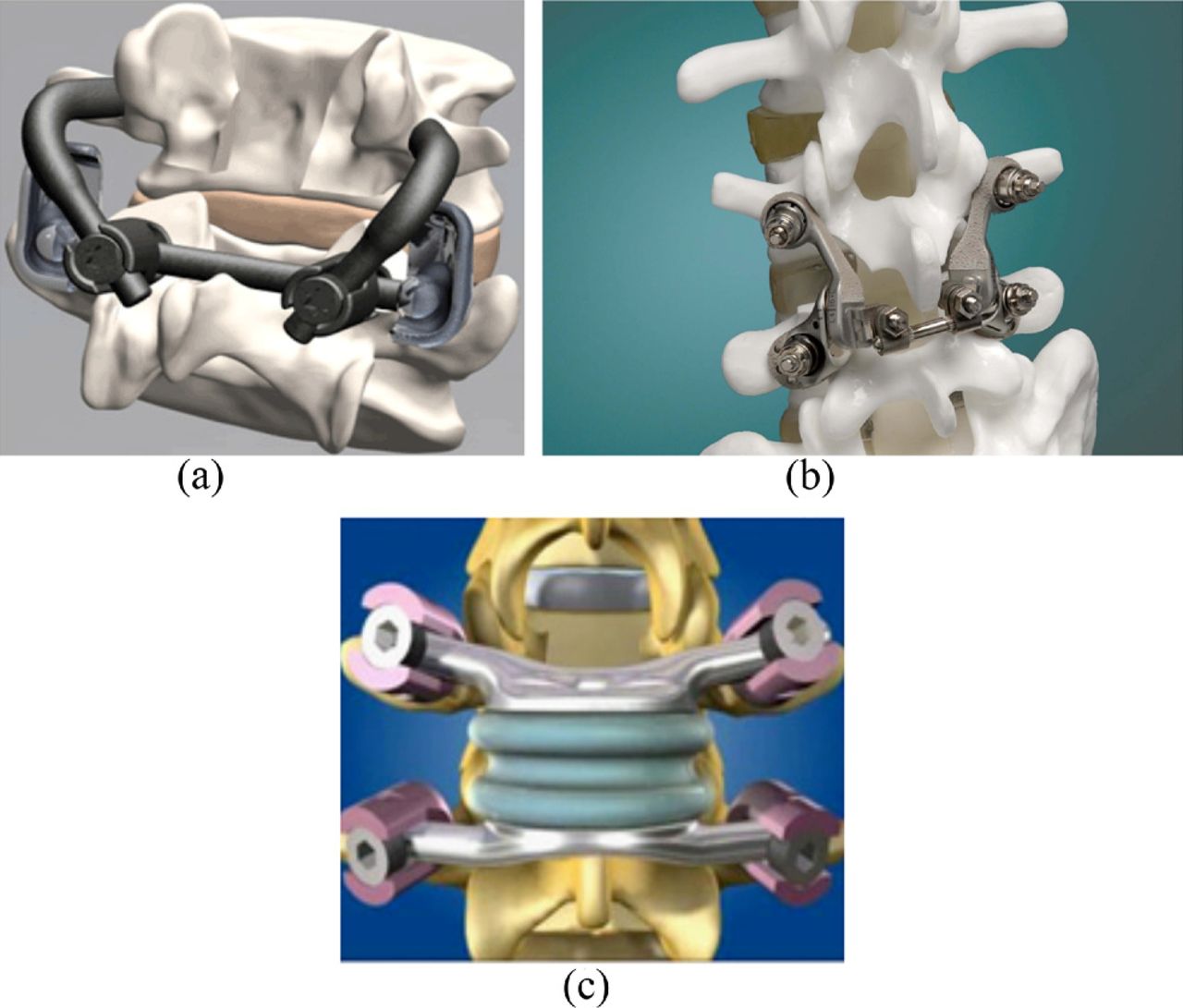
Total facet replacement devices: a, TFAS; b, ACADIA; and c, TOPS.
Facet Solutions offers a device called the ACADIA Facet Replacement System, also called the Anatomic Facet Replacement System (AFRS). It is also pedicle anchor based but includes a metal-on-metal gliding joint that approximates the articular plane of the native facet joints. IDE clinical trials were initiated in 2006, with an estimated study completion date in 2013. In April 2009 the 1-year data for the US Pilot Clinical Study were already published with successful results showing improvements across all measures for lumbar stenosis treatment and no device-related adverse events in any of the patients. The National Health Service (NHS) and National Institutes of Health FDA clinical trial Web site indicate that the IDE study is active, although no verification updates were provided for 2011.
Impliant's system (Princeton, New Jersey) is called the TOPS system. Another pedicle screw– based design, it uses an elastic core that is fixed to the anchors. By its design, the TOPS system would more accurately be categorized as a posterior dynamic fixation device because it functions primarily as a flexible rod. There are not articulating surfaces in the TOPS system. Patients are currently being enrolled in an IDE clinical trial.
Spinal Elements (Carlsbad, California) has borrowed technology from the hand arthroplasty market. Its design is an interpositional resurfacing device that is affixed with a locking bolt that traverses the articular processes. It is distinct from the other devices; it is an articular-surface replacement only, and it does not replace the facet joint and its supporting structures. The TOPS has been in use since 2005, and an FDA trial started in 2006 with completion expected in 2013.
Spinal Elements also offers the Zyre Facet Implant System; it is currently being trialed outside the United States, and this began 2007. It consists of a spacer inserted within the facet joint to augment its natural function while preserving the brunt of the facet joint bony structure.
The future
In the future, facet replacement devices will require a substantial amount of validation testing and numerous clinical studies before they can be considered a viable treatment option for the treatment of spinal disorders. To date, most pathophysiologic research and thus surgical treatments have been focused on the disc as a pain generator. A more comprehensive focus on re-establishing the structure and function of the human functional spinal unit may include facet replacement. A better understanding of facet function and facet-mediated pain, possibly through classification of facet degeneration, may be needed to support the use of such devices.
This approach can be illustrated somewhat by the Functional Spinal Unit (FSU) Total Spinal Segment Replacement device that has recently been released by Flexuspine (Pittsburgh, Pennsylvania) (Fig. 9). The design aims to replace the entire functional spinal unit by an interbody disc component (a metal-on-metal cobalt-chromium core made of articulating paired halves that are inserted through a bilateral Transfacet Lumbar Interbody Fusion [TLIF] approach) and a posterior dynamic resistance component (a sliding rod assembly with silicone dampeners fixed to the spine with pedicle screws). In 2010 the FDA granted conditional approval to begin the initial phase of an IDE study; patient enrollment has yet to start to provide clinical out-come feedback.

Full functional spinal unit replacement device: Flexuspine FSU.
Conclusions
The development of a wide array of motion-preserving devices heralds entrance into an era of partial and complete functional spinal unit replacement. With this, a shift toward using motion-sparing technology instead of fusion-type procedures will most likely take place in the near future. How-ever, indications for these new technologies are still broad and unproven and should be carefully considered and scrutinized, along with efforts to understand pain origins. Prospective randomized clinical studies are essential to prove the safety and efficacy of these technologies and to support our quest to establish and practice evidence-based medicine.
- © 2011 SAS - The International Society for the Advancement of Spine Surgery. Published by Elsevier Inc. All rights reserved.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.