


Abstract
Background Sacroiliac joint (SIJ) dysfunction is an underdiagnosed condition. Several published cohorts have reported favorable mid-term outcomes after SIJ fusion using titanium implants placed across the SIJ. Herein we report long-term (24-month) results from a prospective multicenter clinical trial.
Methods One hundred and seventy-two subjects at 26 US sites with SI joint dysfunction were enrolled and underwent minimally invasive SI joint fusion with triangular titanium implants. Subjects underwent structured assessments preoperatively and at 1, 3, 6, 12, 18 and 24 months postoperatively, including SIJ pain ratings (0-100 visual analog scale), Oswestry Disability Index (ODI), Short Form-36 (SF-36), EuroQOL-5D (EQ-5D), and patient satisfaction. Adverse events were collected throughout follow-up. All participating patients underwent a high-resolution pelvic CT scan at 1 year.
Results Mean subject age was 50.9 years and 69.8% were women. SIJ pain was present for an average of 5.1 years prior to surgical treatment. SIJ pain decreased from 79.8 at baseline to 30.4 at 12 months and remained low at 26.0 at 24 months (p<.0001 for change from baseline). ODI decreased from 55.2 at baseline to 31.5 at 12 months and remained low at 30.9 at 24 months (p<.0001 for change from baseline). Quality of life (SF-36 and EQ-5D) improvements seen at 12 months were sustained at 24 months. The proportion of subjects taking opioids for SIJ or low back pain decreased from 76.2% at baseline to 55.0% at 24 months (p <.0001). To date, 8 subjects (4.7%) have undergone one or more revision SIJ surgeries. 7 device-related adverse events occurred. CT scan at one year showed a high rate (97%) of bone adherence to at least 2 implants on both the iliac and sacral sides with modest rates of bone growth across the SIJ.
Conclusions In this study of patients with SIJ dysfunction, minimally invasive SI joint fusion using triangular titanium implants showed marked improvements in pain, disability and quality of life at 2 years. Imaging showed that bone apposition to implants was common but radiographic evidence of intraarticular fusion within the joint may take more than 1 year in many patients.
This prospective multicenter clinical trial was approved by local or regional IRBs at each center prior to first patient enrollment. Informed consent with IRB-approved study-specific consent forms was obtained from all patients prior to participation.
- sacroiliac joint dysfunction
- sacroiliac joint fusion
- degenerative sacroiliitis
- sacroiliac joint disruptions
- multicenter clinical trial
Background
Pain emanating from the sacroiliac joint (SI joint) may explain 15-23% of chronic low back pain in the outpatient setting.1, 2 SI joint dysfunction (i.e., pain and disability resulting from abnormal function of the joint) may be even more prevalent (up to 40%) in patients with prior lumbar fusion.3, 4 The impact of SI joint pain on quality of life is substantial and often disabling, similar to that observed with other prominent orthopedic conditions such as lumbar spinal stenosis and degenerative hip arthritis.5 Quality of life may be as or more depressed in preoperative SI joint fusion patients compared to those undergoing commonly performed lumbar spine surgeries.6
Substantial evidence points to the SI joint as a valid cause of pain. The SI joint is a richly innervated joint7 and instillation of local anesthetic into the SI joint blocks pain provoked by joint pressurization.8, 9 Therapeutic evidence validates the SI joint as a cause of pain: blinded trials of both periarticular steroids10, 11 and radiofrequency (RF) ablation of the lateral branches of sacral nerve roots have shown (albeit temporary) pain relief.12, 13
Commonly provided non-surgical treatments for SI joint pain include physical therapy, chiropractic manipulations, intraarticular SI joint steroid injections,10, 11, 14 and RF neurotomy (ablation) of the dorsal ramus of L5 as well as the S1-S3 dorsal rami innervating the SI joint.12, 13 But for one 12-month study,15 no high-quality evidence supports long-term pain relief from RF ablation, no study supports long-term pain relief associated with steroid injections, and no published high-quality evidence supports the effectiveness of physical therapy in SI joint pain unrelated to pregnancy.
Open SI joint arthrodesis, which was first reported in the 1920s,16–18 is now infrequently performed for chronic non-traumatic pain, due primarily to relatively large incisions, lengthy hospital stays (3-5 days) and long recovery periods (often lasting months). Complication rates are high,19 non-union relatively common20–22 and patient satisfaction variable.19 Minimally invasive alternatives to open SI joint fusion have gained popularity in recent years23 and most published reports describe use of a series of triangular titanium implants coated with a porous titanium plasma spray (iFuse Implant System®, SI-BONE, Inc., San Jose, CA, USA).24–30 Previously we reported 1-year outcomes from a prospective, multicenter clinical trial of this device in SI joint dysfunction due to degeneration and/or disruption of the joint.31 Herein we report 24-month clinical and radiographic outcomes.
Methods
Sacroiliac Joint Fusion with iFuse Implant System (SIFI, NCT01640353) is a prospective, multicenter single-arm clinical trial. The study protocol was Institutional Review Board (IRB)-approved at each participating clinical site prior to patient enrollment. The study was sponsored by the device's manufacturer (SI-BONE, Inc., San Jose, CA, USA). All study data underwent both remote and on-site data monitoring, and all study data were 100% source-verified.
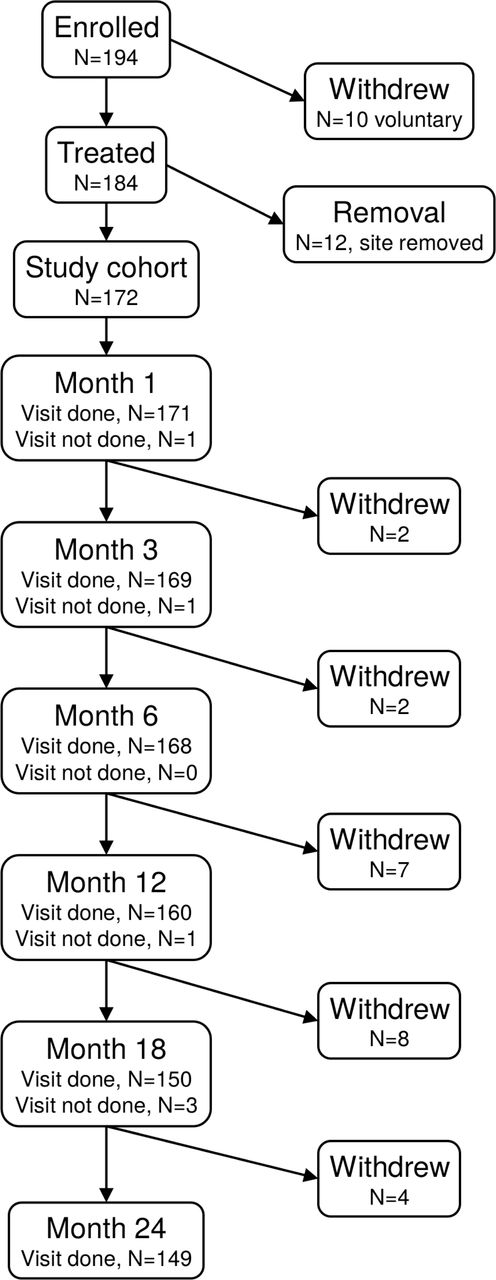
One hundred ninety-four patients were enrolled between August 2012 and December 2013 at 26 sites. Of these, 10 withdrew prior to SI joint fusion and data from 12 subjects at a single site were eliminated due to the site's persistent non-compliance with the study protocol, leaving 172 subjects enrolled and treated. Two additional sites were terminated more than 1 year into the study for protocol non-compliance, resulting in 3 additional subjects not having 24-month study follow-up.
Enrollment criteria were as follows. To participate, adult (age 21-70) patients had to have low back pain for at least 6 months inadequately responsive to conservative care, a baseline SI joint pain score of at least 50 on the 0-100 mm visual analog scale (VAS), an Oswestry Disability Index score of at least 30%, and diagnosed SI joint dysfunction due to degenerative sacroiliitis or sacroiliac joint disruption. Diagnosis was based on a history of pain at or near the SI joint,8 positive provocative testing on at least 3 of 5 established physical examination tests,32 and at least a 50% decrease in pain after image-guided injection/arthrogram into the SI joint with local anesthetic.33–37 Patients had to have the necessary mental capacity to participate, be physically able to comply with study protocol requirements and sign a study-specific informed consent form. Patients were excluded for the following reasons: severe back pain due to other causes (e.g., known hip or spine conditions), diagnosed sacral pathology of other origin, recent (<1 year) major trauma to the pelvis, metabolic bone disease (osteoporosis or other bone conditions), any chronic rheumatologic condition or chondropathy, allergy to titanium, use of medications that impair bone quality or soft-tissue healing, neurologic condition that would interfere with physical therapy, infection, pregnancy or planning to become pregnant, known or suspected drug abuse, psychiatric condition that could interfere with study participation, currently a prisoner or ward of the state, participation in another investigational study or involvement in litigation, on disability leave, or receiving workers’ compensation related to their back or SI joint pain. All study sites obtained IRB approval for the study and patients who agreed to participate signed a study-specific informed consent form prior to any study-specific procedure.
Baseline (pre-surgery) assessments included a detailed medical history, physical examination, and several assessments, including VAS SI joint and lower back pain, disability as measured by Oswestry Disability Index (ODI),38 and quality of life as measured by both EuroQoL-5D (EQ-5D)39 and Short Form-36 (SF-36).40 ODI is a validated ten-question survey for disability due to back pain. EQ-5D is a validated five-question broad quality of life measure that can be combined into a single index and represents the time trade-off (TTO) utility of current health using US norms. EQ-5D US norms (means and quantiles) were taken from population-based surveys.41 SF-36 is a validated 36-question 8-subscaled generic quality of life measure. SF-36 physical component summary (PCS) summarizes overall physical health, with population norms with mean 50 and standard deviation of 10. Similarly, SF-36 mental component summary (MCS) summarizes overall mental health, with similar population norms.
Subjects underwent minimally invasive SI joint fusion under general anesthesia and fluoroscopic guidance, with placement of (typically three) implants, as described previously.31 The iFuse implant, made from titanium with a titanium plasma spray (TPS) porous coating, is triangular in cross section, ranges from 30-70 mm in length and 4-7 mm in inscribed diameter. The implant's triangular shape is designed to minimize rotation and maximize surface area. The procedure incorporates an interference fit between the implant and adjacent osseous walls to reduce micromotion. The porous TPS coating allows for biological fixation in bone. The study protocol asked participating surgeons to perform the procedure according to the manufacturer recommendations, including placement of at least 2 implants across the SI joint. Subjects requiring treatment of both SI joints could undergo either bilateral same-day surgery or staged surgery within 60 days of the first. Subjects were discharged home at the surgeon's discretion. Intraoperative and postoperative measures included number and size of implants, procedure time, fluoroscopy time, blood loss and length of stay.
Postoperatively, subjects were asked to remain at heel-toe touch-down protected weight-bearing using a walker or crutches for three weeks followed by progressive increases in weight-bearing until fully ambulatory. Beginning 1-3 weeks postoperatively, subjects were asked to undergo individualized physical therapy at a recommended frequency of twice a week for 6 weeks. Post-operative physical therapy involved activity modification to minimize pain recurrence, mobility and stability exercises, as well as adjacent segment joint mobilization for stiffness and pain control, and general conditioning exercises. Direct manipulation of the treated SI joint was discouraged.
Subjects were asked to return for study-required follow-up visits at 1, 3, 6, 12, 18 and 24 months post-operatively. Follow-up assessments consisted of review of adverse changes in health, ambulatory and work status, medication use for SI joint or back pain, physical examination, pain, function and quality of life questionnaires. Subjects underwent pelvic lateral, inlet and outlet X-ray at 3, 6 and 24 months and CT scan at 12 months as part of the study.
Adverse events, defined as any negative change in health according to an international clinical trial standard (ISO14155:2011), were monitored continuously and assessed at all study visits. For each event, investigators were asked to rate severity and relatedness to the study device, the device placement procedure and, if present, pre-existing conditions. Relatedness was captured as definitely, probably, possibly, unlikely and unrelated to the device, procedure or pre-existing condition, and each event was categorized by body system.
Study endpoints and cohorts. The primary study endpoint, evaluated at six months after the most recent SI joint fusion (to accommodate subjects who underwent planned staged bilateral surgery), was a binary success/failure composite endpoint. A subject was considered a success if all of the following were met: reduction from baseline VAS SI joint pain by at least 20 points, absence of device-related serious adverse events, absence of neurological worsening related to the sacral spine, and absence of surgical re-intervention (removal, revision, reoperation, or supplemental fixation) for SI joint pain. The 20-point VAS threshold was selected as the minimum clinically important difference (MCID) in chronic lower back pain.42, 43 Various MCIDs for ODI in low back pain have been reported;42, 44–46 we selected a relatively conservative and commonly used threshold of 15. An intent-to-treat approach was used for the primary endpoint; success rates were calculated at subsequent time points using all available data but without imputing missing values. The impact of missing data on pain reduction and ODI was evaluated using a last observation carry forward (LOCF) approach. All other analyses used available data only. The study's secondary endpoints included an analysis of patient success rates at other time points as well as improvement from baseline in VAS SIJ Pain, ODI, SF-36 PCS and EQ-5D scores. All analyses included all enrolled subjects, including a small number of subjects who, during post-enrollment monitoring, were determined to be ineligible for the study. Pre-specified were the following subgroup analyses: underlying condition (degenerative sacroiliitis vs. sacroiliac joint disruption), history of prior lumbar fusion, smokers vs. non-smokers, and unilateral vs. bilateral SI joint fusion. Additional unplanned subgroup analysis was performed.
The study included detailed imaging analyses of 3, 6, and 24-month X-rays as well as a 12-month high-resolution pelvic CT scan. All CT scans were read independently by 3 non-conflicted bone radiologists subcontracted by an independent core radiographic laboratory (BioMedical Systems, Inc. St. Louis, MO). A “2 out of 3” approach was used to determine a consensus read for each endpoint, with the main reader adjudicating discrepancies. The primary imaging endpoint was the proportion of subjects showing at least 30% apposition of bone to both the iliac and sacral sides of at least 2 of 3 iFuse implants on 12-month CT scan. Primary safety parameters included: radiolucency (defined as lucency around the device consistent with possible loosening of the device), device failure/fracture, device breach (defined as presence of the distal end of the implant in the sacral foramen or outside the osseous envelope of the sacrum), device migration and adverse bone reaction. Additional radiographic endpoints included bridging bone across the SI joint (and extent thereof), positive bone remodeling response and heterotopic ossification. A single reader read inlet/outlet/lateral pelvic X-rays at months 3, 6 and 24, scoring a subset of the above-described endpoints.
Sample Size. Study sample size was determined by pre-planned interim analysis with a predictive enrollment stopping rule. Study enrollment was stopped in December 2013 with 172 subjects enrolled and treated as a result of a positive predicted success rate analysis.
Statistical analysis. For continuous variables, changes from baseline were compared using repeated measure analysis of variance. Confidence intervals for proportions were calculated using standard methods. Analysis of procedure-related variables focused on the index (first side) procedure only. All statistical analyses were performed using R.47
Results
Of 194 patients who were eligible and signed a consent form, 10 voluntarily withdrew prior to SI joint fusion. All data from 12 subjects at a single site were excluded due to persistent non-compliance with the study protocol, leaving 172 enrolled and treated subjects. Analysis excludes data from an additional 3 subjects, who were excluded after the month 12 visit for persistent site non-compliance with the protocol. During post-enrollment monitoring, 21 patients (Supplementary Table 1) were found to not meet all eligibility criteria; as these subjects were already enrolled and treated, all were included in statistical analyses.
Patients included but who did not meet all study eligibility requirements.
Baseline characteristics. Subjects averaged 50.9 years old, most (96.5%) were Caucasian and 69.8% were women. Baseline SI joint pain (mean 79.8 points, 0-100 scale) and Oswestry Disability Index (ODI, mean 55.2) levels were high. On average, pain had persisted for 5.1 years (range 0.43 to 41.08 years). Quality of life (QOL) was substantially diminished (mean EQ-5D TTO of 0.43 and mean SF-36 PCS of 31.7), values that are substantially lower than normal populations.5 131 (76.2%) subjects were taking opioid medications for SI joint or low back pain at baseline and many subjects (44.2%) had a history of prior lumbar fusion. SI joint pain persisted despite prior SI joint-focused physical therapy (64.5% of subjects), prior SI joint steroid injections (94.2%) and prior RF ablation of the SI joint (15.7%).
Procedure characteristics. Fourteen (8.1%) subjects underwent planned bilateral SI joint fusion. Procedure time averaged 46.6 minutes (range 13.0 to 111.0 minutes) and mean estimated blood loss was 51.0 cc (median 30 cc, range 5.0-800.0 cc, see Table 2). One subject, an outlier, had 800 cc of blood loss due to injury to the superior gluteal artery. Four implants were used in 12.8% cases; three were used in 83.7% of cases and 2 in 3.5% of cases. Subjects were discharged in a mean of 0.79 days (range 0 to 7 days); 164 (95.3%) were discharged either the same day or within 2 days.
Characteristics of enrolled subjects.
Index procedure characteristics (n=172). Only the index side procedure is reported.
Subject trial flow. Of the 172 participants, 167 (97.1%) had 6-month follow-up, 157 (91.3%) had 12-month follow-up and 149 (86.6%) had 24-month follow-up (Figure 1). Reasons for incomplete 24-month follow-up included the following: 5 had withdrawn consent to participate, 2 had died from causes unrelated to the SI joint, 10 were documented to be lost to follow-up, 5 were unavailable for other reasons, including termination of the site by the sponsor in 3 subjects and substance abuse in 1.

Patient study flow.
Primary endpoint. At month 6, 138 of 172 subjects met the study's success endpoint definition, for an intent-to-treat success rate of 80.2% (95% posterior credible interval 73.8-85.7%). Using available data only, the 12-month success rate was 127/159 (79.9%) and the 24-month success rate was 119/149 (79.9%). At all time points, the observed success rates exceeded the study's pre-determined threshold for success (Bayesian posterior probability of study success >0.999). Pre-specified subgroup analysis showed no statistically significant differences in 24-month success rates by underlying diagnosis, a history of prior lumbar fusion, smoking status, or unilateral vs. bilateral SI joint fusion surgery.
Pain and quality of life outcomes. During follow-up, mean (SD) SI joint pain improved from a baseline of 79.8 (12.8) points to 26.0 (26.7) points at month 24 (see Figure 2); the reduction in pain was statistically significant (p<.0001) at every time point, including month 1. Similarly, mean (SD) Oswestry Disability Index score, a measurement of disability due to back pain, improved from a baseline of 55.2 (11.5) points to 30.9 (20.5) at month 24, with statistically significant reductions at each time point. For both VAS SI joint pain and ODI, there was no variation in response by preplanned subgroups (underlying diagnosis, history of prior lumbar fusion, smoking status, or bilateral vs. unilateral procedures). Additional subgroup analyses (e.g., age, race, ethnicity, number of postoperative rehabilitation visits, gender, years of SI joint pain, history of pain beginning during pregnancy or soon thereafter) showed no variation in response. Analyses that included imputation of missing data using the LOCF method showed only minimal differences in 24-month SI joint pain and ODI reductions resulting from missing data (-3.1 points [0-100 scale] for VAS SI joint pain and -1.6% for ODI [0-100% scale]). The proportions of subjects having VAS SI joint pain improvements ≥20 points at 6, 12 and 24 months were 82.2%, 81.8% and 83.9%, respectively. The proportions having ODI improvements ≥15 points were 65.7%, 66.7% and 69.1%, respectively. The proportions of subjects with SI joint pain improvements of at least 40 points at months 6, 12 and 24 were 66%, 70% and 72%, respectively. The proportions with ODI scores <30 (indicating no or mild disability) were 46.2%, 50.3% and 46.3%, respectively. That is, the ODI improvement seen at 6 months was maintained at 2 years.
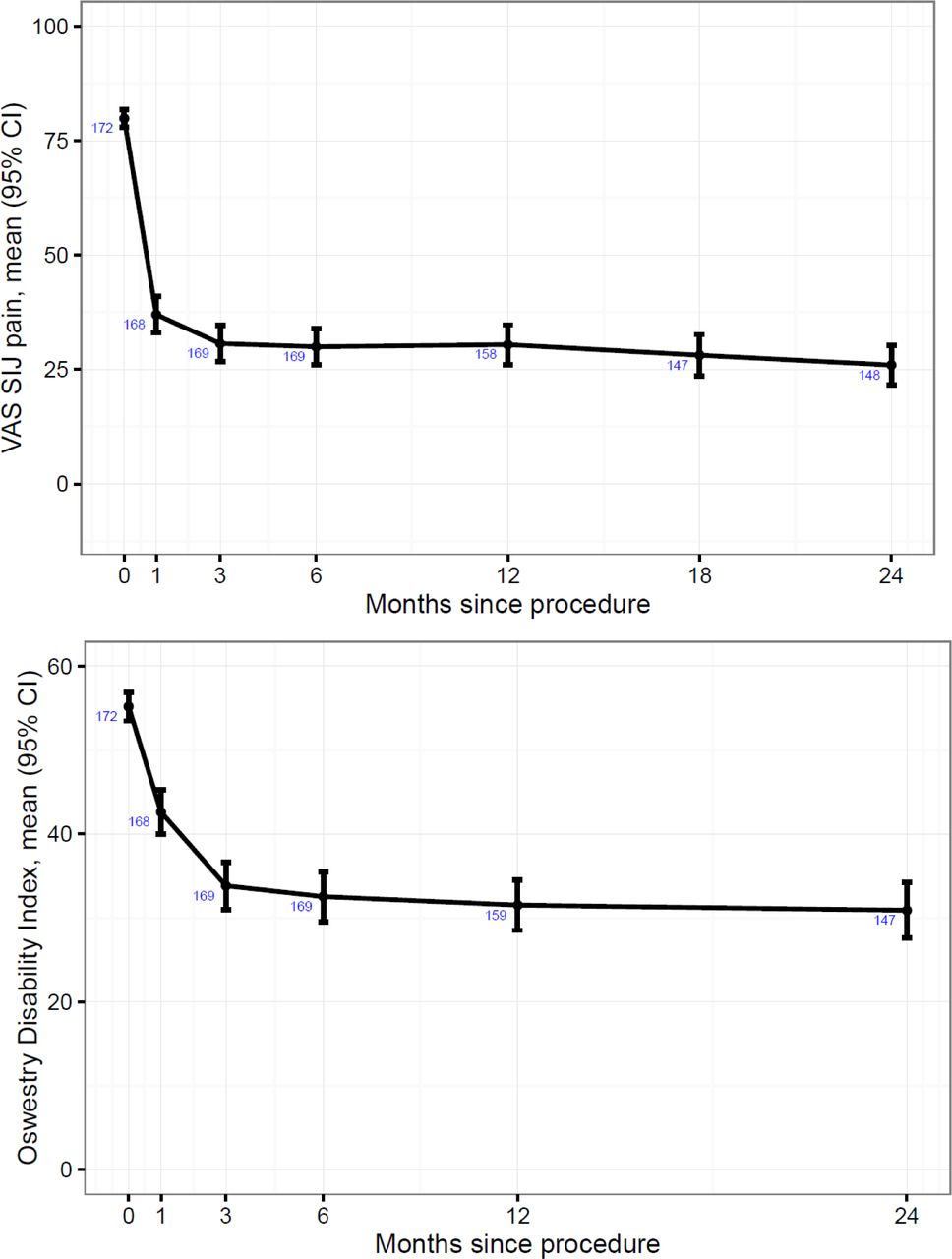
Improvement in VAS SI joint pain (top) and Oswestry Disability Index (bottom). Numbers in blue indicate the number of subjects assessed. ODI was not assessed at month 18.
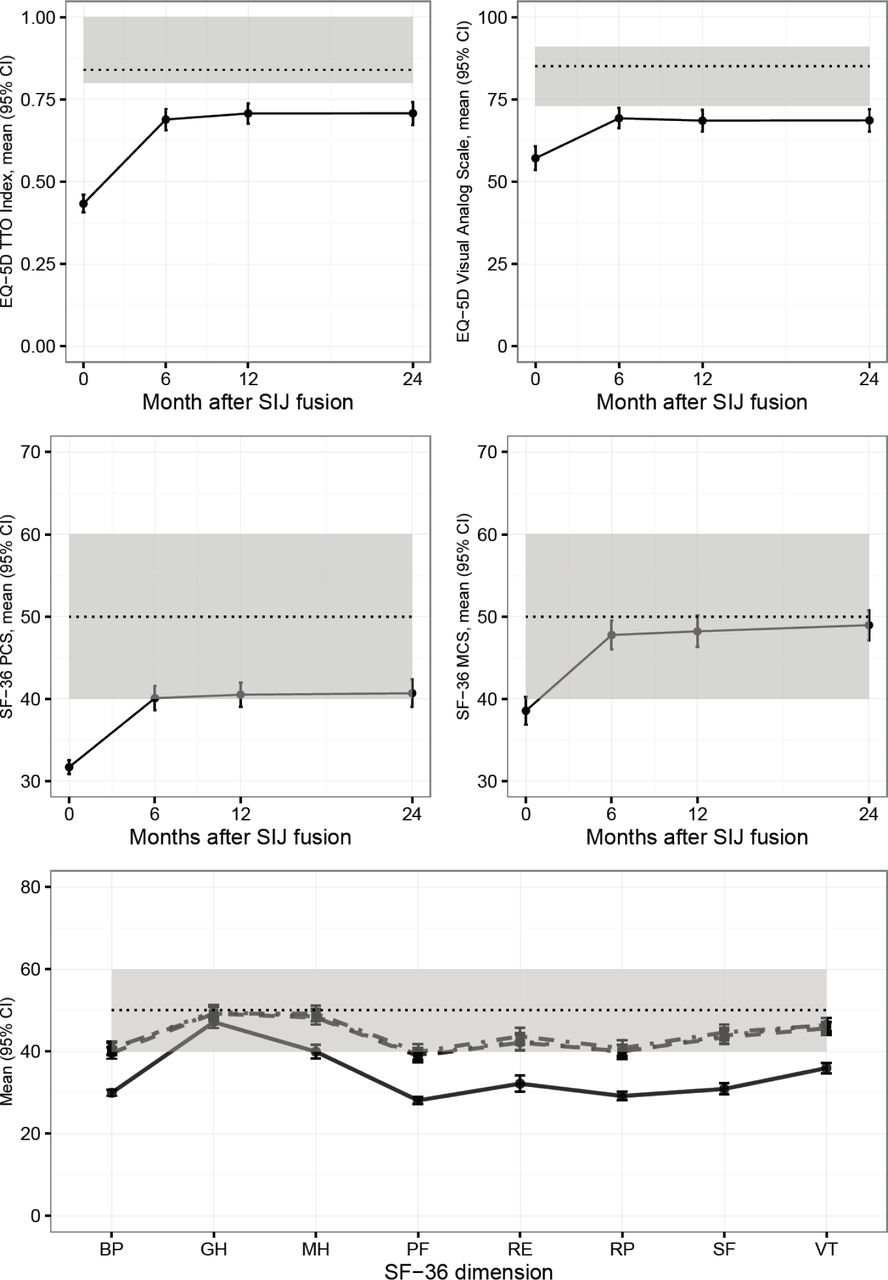
Quality of life was measured using EuroQOL-5D and SF-36, two commonly used global assessments. Over the 24-month follow-up period, EQ-5D time trade-off index, which was markedly depressed at baseline (mean 0.43) improved by 0.27 points (p<.0001, Table 3). Similarly, compared to baseline, all SF-36 subdomains improved at all time points assessed (Figure 3).

Changes in EQ-5D time trade-off index and visual analog scale (top row), SF-36 PCS and MCS (middle row) and SF-36 individual dimensions (norm-based scale). Dotted lines show population medians (top row) or means (middle and bottom row) and 25th/75th percentiles (top row), 1 SD (middle and bottom row). In the bottom row, solid=baseline, dashed= 6 months, dotted=12 months, dot-dashed=24 months.
Change over time in SI joint pain, Oswestry Disability Index, SF-36, and EQ-5D.
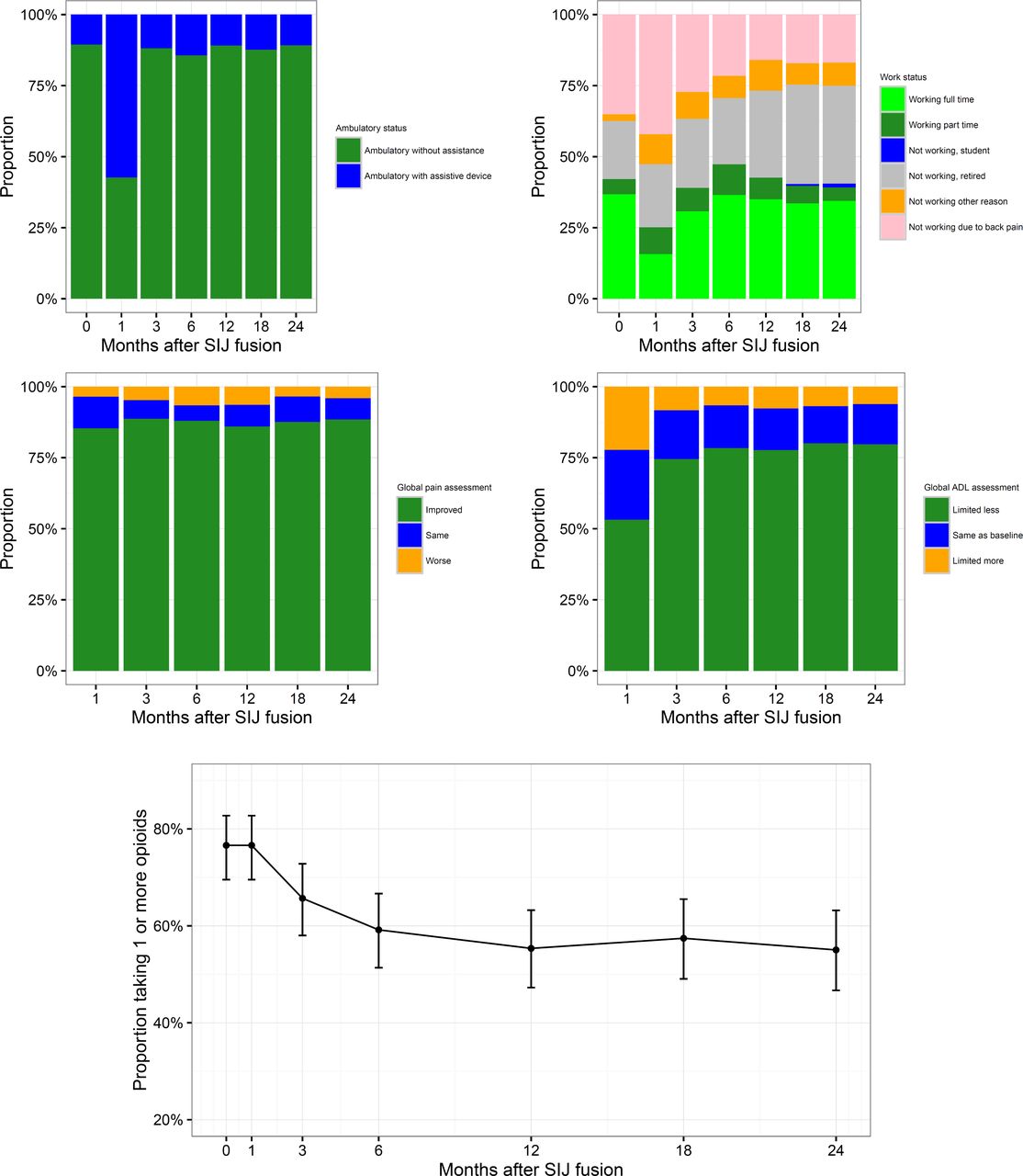
Other effectiveness outcomes. At each study visit, subjects were asked to report ambulatory and work status, and to compare current pain levels and ability to perform activities of daily living (ADLs) to their status prior to surgery (Figure 4). Prior to SI joint fusion, 89.5% of subjects were fully ambulatory. Recovery of loss of ambulatory status related to surgery was rapid (by month 3, Figure 4) and by month 24, the same proportion (89.2%) reported being fully ambulatory. Prior to SI joint fusion, 37.4% of subjects were not working due to back or other pain. By month 24, the proportion not working due to back or other pain decreased to 25.0% (McNemar test, p=0.0013). Throughout follow-up, subjects reported improved pain compared to baseline, with 88.5% reporting decreased pain at 24 months and only 4.1% reporting an increase in SI joint pain. Recovery of activities of daily living was also rapid: one month after surgery 53.2% reported less limitations of ADLs, and by month 24, 79.7% reported less limitation and only 6.1% reported more limitation.
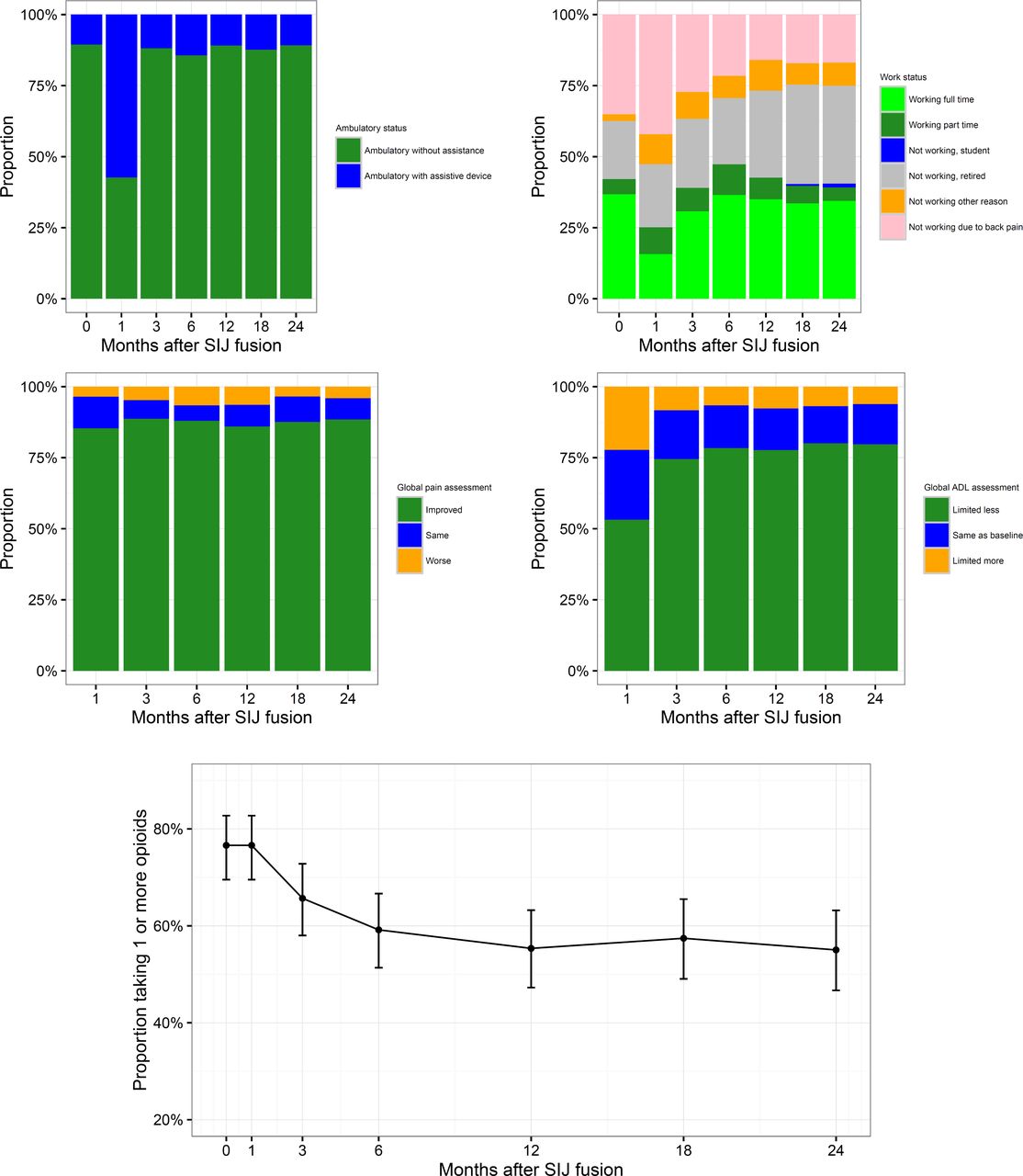
Changes by month in ambulatory status (top left), work status (top right), global pain assessment (middle left), activities of daily living (middle right) and opioid use (bottom, proportion ± 95% confidence interval).
Satisfaction rates were high, with 78.1% reporting being very satisfied with SI joint treatment by month 24 and 93.8% being very or somewhat satisfied. 74.7% indicated they would definitely have the procedure again; 88.4% indicated they would probably or definitely have the procedure again.
Medication use. At each study visit, medications used for SI joint or low back pain were collected. The proportion of subjects taking opioid analgesics for SI joint or low back pain decreased from 76.2% immediately prior to surgery to 55.0% at month 24 (Figure 4). At month 24, 37 who were taking opioids at baseline had stopped taking them; in contrast, only 7 who were not taking opioids at baseline had started them by month 24 (McNemar test, p<.0001). Of those who began taking opioids, 2 had had contralateral SI joint fusion, one had recently undergone revision surgery, and 1 had back pain related to a fall 5 months after SI joint fusion.
Device-related events. All negative changes in health were collected as adverse events, consistent with an international clinical trial standard (ISO 14155:2011). The number of adverse events was large (454 events in 153 subjects), but the majority were unrelated to the device or SI joint fusion procedure (a full listing of adverse events is provided in Supplementary Table 2). Four adverse events (2.4% of all subjects) were rated by the investigator to be definitely device-related (Table 4) and 3 (1.8%) were probably related. Neuropathic pain related to implant impingement on sacral nerve roots occurred in 3 cases (including one non-study-related case), all of which resolved with immediate repositioning of implants. In 4 cases, SI joint or hip pain was attributed to the presence of an implant or bone growth around the implant.
Listing of adverse events by frequency.
Device- or procedure-related events.
Procedure-related events. Twenty-six events were rated as probably or definitely related to the placement procedure (Table 4). The most common events were wound infection, irritation or drainage, SI joint pain related to implant malposition (described) above, and recurrent SI joint pain related to inadequate device placement. One subject had a deep wound infection that required surgical debridement.
Adverse event severity. Seventy-three events were noted to be severe, of which 1 was probably or definitely related to the device and 7 were probably or definitely related to the procedure. The device-related severe event was the above-described case of neuropathic pain related to suboptimal implant placement. The 6 procedure-related events were the above case plus: two cases of recurrent or persistent pain due to suboptimal implant position requiring revision surgery (described above), one case of postoperative surgical pain requiring brief hospitalization, one case of postoperative nausea/vomiting requiring prolonged hospitalization, and one case of deep wound infection requiring surgical wound debridement.
Revisions. Revision surgery of the index side(s) occurred in 8 cases (4.7%). In 2 cases, subjects awoke with new onset leg pain; pain resolved when implants were repositioned slightly. In 4 cases, SI joint pain improved only minimally after SI joint fusion and CT scan showed suboptimal implant placement, with the lower implants not placed sufficiently into the sacrum. In 1 case, radiolucencies along the implants were present on CT scan. All 4 underwent placement of additional devices with improvement in SI joint pain thereafter. One subject underwent staged bilateral SI joint fusion with initial pain relief on the index side followed by pain recurrence 6 months later. Workup showed bilateral labral tears and evidence of possible femoral acetabular impingement. Since repeat bilateral SI joint block provided temporary pain relief but intra-articular hip injections did not, he underwent traditional open SI joint arthrodesis followed by placement of one additional implant in each SI joint, which resulted in improved SI joint pain. Finally, one subject underwent L4-S1 fusion at a different institution approximately 13 months after right-sided SI joint fusion. A few months later, recurrent indexside SI joint pain developed. CT showed that the S1 screw was touching the proximal SI joint implant. Because the subject had pain relief with a repeat SI joint block, the subject underwent revision surgery in which the caudal implant was removed and an additional (non-iFuse) device placed across the joint. The investigator believed that recurrent SI joint pain could have been from additional stress transfer to the SI joint after the lumbar fusion, or possibly related to the S1 pedicle screw touching the proximal iFuse implant.
Imaging findings. Imaging analysis was based on scheduled pelvic X-rays (inlet, outlet and lateral) at months 3, 6 and 24 and a 12-month high resolution pelvic CT scan. The study's primary radiographic endpoint was based on the 12-month CT scan.
X-ray analysis. 146 subjects (84.9%) underwent 1 or more scheduled pelvic X-rays (inlet, outlet and lateral) at months 3, 6 and 24. Of these, 128 (87.7%) had an X-ray at all 3 time points. Areas of lucency around one or more implants was present on the right side in 39 subjects and on the left side in 33 subjects. In the majority of cases (24 and 21 subjects, respectively) radiolucency was confined to the distal tip of the implant within the sacrum. In a minority of cases (15 and 12 subjects, respectively), radiolucency on the right or left side spanned a longer section of the implant within either the sacrum or ilium (Table 6).
CT analysis. Twelve month CT was done in 159 of 161 (98.8%) subjects still participating at month 12. Bone adherent/adjacent to placed implants covering >30% of the surface area of the implant was seen in >90% of implants (Table 5). Bony apposition to at least 30% of the implant surface area on both the iliac and sacral sides of 2 or more implants, the study's primary imaging endpoint, occurred in 97% of treated sides. Of the 3 cases where apposition could not be confirmed, the CT was technically inadequate in 2. Radiolucency was seen in 9 of 91 cases on the right and in 8 of 93 cases on the left. The degree of radiolucency was <15% in most cases and radiolucency was typically seen when the distal end of the implant was barely (e.g., <1 cm) into the sacrum. Breach of the implant into the sacral foramen was present in a small number of cases (4 on the right and 8 on the left). No patient had symptoms related to breach seen on the month 12 CT. No device failure or device migration was seen. Adverse bone reaction was generally absent; in 3 (right) and 6 (left) cases, small cystic changes or scattered erosions were seen. Bone remodeling was seen in >80% of treated SI joints. Bridging bone was seen in a minority of cases (20 on the right, 19 on the left) either adjacent to or distant from placed implants.
Findings from CT analysis.
Findings from X-ray analysis.
Discussion
Our study, the first prospective multicenter report of 2-year outcomes after minimally invasive SI joint fusion, provides strong evidence that patients can be diagnosed with SI joint dysfunction and successfully treated with triangular titanium implants placed during a minimally invasive surgical procedure. Study data show marked improvement in SI joint pain extending to 2 years, with parallel changes in disability as measured by ODI, and quality of life as measured by two commonly used instruments (SF-36 and EQ-5D). The proportion of subjects with clinically important (≥15 points) improvement in ODI was meaningful (about two-thirds). Quality of life improvements were substantial as measured by EQ-5D and approximately 1 SD for SF-36. We observed parallel improvements in self-rated global assessments of pain levels, limitations in activities of daily living, and high rates of satisfaction and desirability of undergoing the procedure again. Full ambulatory status was preserved in the majority of patients. The proportion of subjects not working due to back pain decreased.
Results from our study are supported by several retrospective case series,24–30 including some with up to 4.5- and 5-year follow-up,49 as well as 12-month results from a prospective randomized trial that showed little response to non-surgical treatment in the same patient population.50 As such, our trial provides strong support for minimally invasive SI joint fusion as a standard treatment choice in patients with SI joint dysfunction unresponsive to non-surgical treatment. Our study also confirms that a minimally invasive surgical approach is associated with expected benefits of minimal blood loss, relatively short procedure times, a short hospital length of stay, and a low rate of adverse events.
SI joint pain has a marked impact on quality of life. Baseline quality of life scores in study subjects are consistent with a high burden of disease5 and quality scores in our cohort were at least as depressed as those of other major orthopedic conditions commonly treated surgically, such as lumbar spinal stenosis and degenerative hip arthritis.5 We observed marked improvements in quality of life after SI joint fusion with two commonly used quality of life instruments (EQ-5D and SF-36). The improvements seen in all measured parameters (pain, disability and quality of life) were impressive given the long duration of SI joint pain (on average 5 years) and the high rate of failure of prior therapies (64% had received physical therapy, 94% had received SI joint steroid injections and 16% had received RF ablation). In contrast to other studies of spine surgeries, in which patients with prior spine surgery are typically excluded, our study allowed patients with prior lumbar fusion to participate, as this is known to be a risk factor for SI joint degeneration,51 possibly by increasing adjacent segment stresses.52 Prior lumbar fusion was common (44.2%) and many patients had a history of concomitant spine and hip disease, making the observed improvement in pain and QOL all the more impressive. Interestingly, pre-specified subgroup analysis showed that subjects with a history of prior lumbar fusion responded similarly to those without such a history.
Outcomes observed in our trial were comparable to those of other accepted surgical spine treatments. In our study, 69.1% of subjects had 15 or more point improvement in ODI at 2 years, values similar to that observed after CHARITE artificial disc replacement (68%53). In an early study of lumbar fusion using recombinant bone morphogenetic protein, 83% of subjects had a 15-point or more improvement in ODI at month 24.54 In another randomized trial of lumbar fusion with recombinant bone morphogenetic protein, 71% had 15-point improvements.55 Mean improvement in ODI in our cohort (24.5 points) was about the same as that observed in SPORT's lumbar degenerative spondylolisthesis study (24 points56), slightly larger than that observed in SPORT's lumbar stenosis trial (20 points in the “as treated” analysis of the randomized and observational cohort57), and smaller than that observed in SPORT's lumbar disc herniation study (37.6 points58). Improvements in our cohort occurred despite the common finding of prior lumbar fusion (44.2%) and concomitant hip or spine disease (24.4% spinal stenosis, 14.0% hip problems, 9.3% prior sacral trauma), conditions that often result in exclusion from other clinical trials.
SI joint pain has often been called “controversial” and challenging to diagnose, especially because other conditions (e.g., lumbar disc disease, hip osteoarthritis) can mimic SI joint pain. Unfortunately, no imaging study finding currently available is pathognomonic of SI joint dysfunction. Similar to other painful conditions, there is no gold standard for the diagnosis of any pain generator, and the idea that SI joint dysfunction diagnosis is challenging may stem, in part, from this void. Nonetheless, the diagnostic algorithm used in our study, which includes a typical history of off-center buttocks or low back pain below L5 and a positive Fortin finger test,59 positive findings on 3 or more physical examination provocative maneuvers that stress the SI joint and reproduce SI joint pain, and confirmatory diagnostic SI joint block, identified a patient population with a high response rate to definitive treatment, with high long-term positive response rates. The high response rate validates the diagnostic approach. The use of diagnostic blocks in other pain conditions is commonly performed, and confirmatory diagnostic SI joint block is recommended by multiple pain and anesthesia specialty societies.33–37
Opioid use in the US has been termed an epidemic60, 61 and concern remains about rising rates of prescribed opioid use for pain treatment. Chronic opioid use after lumbar fusion is common, occurring in >50% of patients in one study.62 In this study, chronic opioid use was associated with less return to work, and chronic opioid use prior to lumbar fusion was associated with failed back syndrome, additional surgery, depression and extended work loss. In a community-based comparison of surgical vs. non-surgical treatments for chronic low back pain, the proportion of patients taking opioids at 1 year decreased in non-surgical but not surgical patients.63 In a prospective case series, the proportion of spine surgery patients who were taking opioids preoperatively and at 1 year did not change.64 In the SPORT studies, the likelihood of opioid use amongst patients taking opioids at baseline decreased, while the incidence of new opioid use amongst non-users at base-line was modest.65 Surgical treatment in SPORT was associated with a lower incidence of opioid use compared to non-surgical treatment, but the non-randomized nature of treatment selection severely limits the comparison. Moreover, the overall proportion of SPORT participants taking any opioids at each study visit was not reported. In our study, the incidence of opioid use at baseline was high (76.2%), which may reflect the underlying severity and chronicity of disease, and the reduction in opioid use was important (55.0% at 24 months, a reduction of 28.2%). This reduction was consistent with results from a randomized trial of SI joint fusion, showing a reduction of opioid use at 6 months in the SI joint fusion group but an increase in the non-surgical group.50 We note that pain and disability did not resolve completely in our cohort, and the use of opioids remained high. Potential explanations include: 1) the presence of comorbid conditions that require opioid use, 2) the long duration of SI joint pain (average of 5 years in our cohort), which could itself limit pain and disability improvement due to social or psychological factors, and 3) complications from prior surgeries, especially lumbar fusion, which may have been provided to patients on the basis of misdiagnosis of lower back pain as coming from the spine instead of the SI joint.
Imaging findings in our study, read by radiologists at an independent core laboratory, were instructive. The titanium implants used to perform SI joint fusion were designed for biological fixation in bone, and the study's primary imaging endpoint, the proportion of subjects showing bony apposition to at least 30% of the implant surface area on both the iliac and sacral sides of 2 or more implants, occurred in 97% of treated sides, validating the implant's design.
The presence of bone on the implants suggests good bony integration and permanent stabilization of the SI joint. The number of implants with radiolucencies suggestive of implant loosening were small. As noted above, 97% of patients showed adherence of bone to at least 2 implants on both the sacral and ilial sides. The significance of scattered radiolucencies on implants is therefore unclear. Lucencies were more apparent on X-ray; these were typically around the leading edge of the implants that were placed less than 1 cm deep into the target sacrum. While lucencies along the implant's length may suggest a risk for inadequate fixation, the clinical impact may be limited in the long term by fusion across the SI joint itself. To date, only 1 of 172 patients underwent implant revision as a result of symptomatic loosening that was apparent along a major portion of the implant. Implant breach into the sacral foramen or beyond the sacral cortical bone anteriorly or posteriorly were present in a small number of patients, most of whom were asymptomatic. No device failures or migration were seen. Adverse bone reaction was generally absent, with a small number of cases showing small cystic changes or scattered erosions. Bone remodeling, predominantly in the ilium, was seen in >80% of treated SI joints, suggesting a positive impact of stabilization on bone structure.
Bridging bone across the SI joint was seen in a minority (approximately 22%) of cases. Given that a five-year study using the same imaging technique (pelvic CT) showed bridging bone in most cases (87%),49 it is apparent that the technique used in our study may require longer periods of time to produce radiographically visible full bony bridging across the joint. Whether alternative techniques aimed at accelerating bony fusion across the SI joint (e.g., intraoperative decortication) produce incremental patient benefits is unknown. What is clear is that both short- and long-term data from our study indicate excellent pain, disability and quality of life improvements with the current device and technique, providing strong evidence that placement of porous TPS-coated triangular titanium implants transarticularly across the SI joint through a lateral approach results in clinically important beneficial patient outcomes. Implants clearly blocked joint motion and produced positive clinical outcomes even without radiographically visible arthrodesis of the SI joint (at 1 year).
In our cohort, revision surgeries were required in a small number of cases. Early revisions were performed in 2 cases to address radicular symptoms related to implant malposition; radicular pain resolved rapidly in both subjects on repositioning of offending devices. In 4 cases, SI joint pain did not improve and CT scan showed poor implant placement with caudal implants not fully engaging the sacrum. Revision surgery in these cases, with placement of additional implants, resulted in improved symptoms. These findings highlight the importance of placing a sufficient number of devices across the SI joint. Multiple studies have demonstrated superior SI joint stability with two-screw constructs compared to a single screw for unstable pelvic fractures.66–68 Moreover, biomechanical studies show that placement of 3 iFuse implants across a SI joint reduced motion in flexion-extension, lateral bending, and axial rotation, with larger reductions observed using a transarticular configuration when compared with an inline (posterior) configuration.69 One subject had pain associated with radiolucencies around the implant. Finally, in one study subject, failure to improve may have been explained by competing pathology. Overall, the revision rate for minimally invasive SI joint fusion appears to be low (3.6% or less at 4 years)70 compared to other lumbar spine surgical procedures.71, 72
All negative changes in health were collected as adverse events in our study, which resulted in a high number of reported events. However, because the study protocol used an international clinical trial standard (ISO14155:2011), which defines an adverse event as any negative change in health, most adverse events were unrelated to the study device or procedure. Only 7 events were deemed related to the study implant, of which 3 were related to poor implant placement causing nerve irritation and one was related to a fall associated with inadequate implant placement. 26 events were deemed related to the study procedure; the events were not unexpected and all resolved.
Several devices are now commercially available in the US and other geographies to perform minimally invasive SI joint fusion. Some of these devices are placed across the articular SI joint using a lateral-to-medial approach, as done in our study. Other devices (none of which are currently cleared by US FDA) are placed into the dorsal ligamentous portion of the SI joint via a posterior approach. The dorsal/posterior approach typically involves removal of a portion of the dense dorsal ligaments and the interosseous ligament, which may acutely destabilize the joint.73, 74 As no implants are placed across the SI joint with the dorsal technique, the biomechanics of the dorsal technique will differ from the lateral technique. Thus, while it is unknown whether the results of our study are applicable to other lateral transfixing devices, it is highly unlikely that our results are applicable to devices placed through a dorsal approach.
Our study has several advantages. First, participants were carefully screened against predetermined eligibility criteria and results represent the prospective experience of multiple surgeons. Second, the trial was executed according to an international clinical trial standard (ISO14155:2011). Third, study data were collected on electronic case report forms at pre-determined postoperative time points and all data were rigorously monitored and source verified.
The study has two primary limitations: 1) lack of a concurrent control group undergoing non-surgical treatment and, 2) a 24-month follow-up rate that was not as high as desired. Regarding the control group, direct comparisons can be made to INSITE (NC-T01681004), a companion, randomized clinical trial run contemporaneously with the current study. In INSITE, patients enrolled using identical eligibility criteria were randomized to receive either SI joint fusion using the same device/procedure as used herein or non-surgical management, consisting of medication management, physical therapy, SI joint steroid injections and radiofrequency ablation of the SI joint. Baseline characteristics of SIFI and INSITE were very similar.50 12-month results from INSITE indicated a profound difference in response between surgical and non-surgical care, with superior outcomes for all effectiveness parameters measured. Moreover, pain and disability responses to SI joint fusion were very similar between the two studies. INSITE's non-surgical group's response validates that the positive health effects seen in the current study are robust and distinct from the natural course of disease. Marked improvement in pain, disability and quality of life in the absence of a definitive intervention would not be expected anyway, given the long mean pain duration (>5 years on average) and the lack of prior response to physical therapy and steroid injections, which were common in the treated patient population and which have very little support in the literature as being effective.
Second, 13.4% of subjects were lost to follow-up during the study or did not have 24-month visits. Pain and ODI scores in exiting subjects were higher than subjects who continued to participate; however, the impact of missing values on pain and ODI scores were analyzed and found to be minor, and do not effect overall study conclusions. Recently published 24-month follow-up rates in spine surgery randomized trials conducted under investigational device exemption have shown similar long-term participation rates (83%/80%,75 89%/82%,76 98%/94%,77 91%/90%,78 81/82%,79) to that in our study (86.6%).
Combined with results from other studies, our study confirms that SI joint fusion with iFuse implants has proven to be safe and effective, and hence a reasonable surgical treatment option for patients with SI joint dysfunction not responsive to non-surgical care.
Conclusions
This prospective multicenter clinical trial provides strong evidence that minimally invasive SI joint fusion using triangular porous coated implants placed across the SI joint improved pain, disability and quality of life at 24 months in patients with SI joint dysfunction due to degenerative sacroiliitis and sacroiliac joint disruption, and was associated with a decrease in opioid usage for SI joint or low back pain. Imaging showed evidence of stabilization and early fusion of the SI joint.
Disclosures & COI
This study was sponsored by SI-BONE, Inc. Bradley Duhon, Fabien Bitan, Don Kovalsky and Harry Lockstadt conduct clinical research for SI-BONE. Bradley Duhon, Fabien Bitan and Harry Lockstadt are paid consultants to SI-BONE. Travis Hillen worked as a subcontractor to BioMedical Systems, Inc., as an independent core laboratory radiographic reader. Daniel Cher is an SI-BONE employee.
Acknowledgements
The authors acknowledge Kathryn Wine, Denise Law, Elaine Wilhite, Terrill Himmelmann, Corinne Lee, Steve Scott, and Jeff Price from SI-BONE for study monitoring, and SI-BONE, Inc., for study sponsorship.
The authors acknowledge the 67 investigators and coordinators in the SIFI Study Group: Harry Lock-stadt, MD, Elaine Wilhite, MS, James Farris, PA-C (Bluegrass Orthopaedics and Hand Care, Lexington, Kentucky); Don Kovalsky, MD, Cristy Newman, Laura Pestka, RN (Orthopaedic Center of Southern Illinois, Mount Vernon, Illinois); Cheng Tao, MD, Jackie Makowski, Toni Kelly (Spine and Neuro Center, Huntsville, Alabama); S. Craig Meyer, MD, Vicki Jones, Michelle Vogt (Columbia Orthopaedic Group, Columbia, Missouri); Scott Kutz, MD, Linda Thompson, RN, BSN, FNP (Mercy Medical Research Center, Springfield, Missouri); Dimitriy Kondrashov, MD, Irina Kondrashov (SF Spine Group, San Francisco, California); Andy J. Redmond, MD, Jennifer Piazza, MS, Laurie Doredant, Beth Short, BSN, MS, Jessica Mayfield, RN (Precision Spine Care, Tyler, Texas); CL Soo, MD, Julie White, MBA, Kallena Haynes (Medical Research International, Oklahoma City, Oklahoma); Bradley Duhon, MD, Amber Pfister (Neurosurgical and Spine Specialists, Parker, Colorado); Ali Mesiwala, MD, Stephanie Bose, RN (Southern California Center for Neuroscience and Spine, Pomona, California); Leonard Rudolf, MD, John Thibodeau Jr RN (Alice Peck Day Memorial Hospital, Lebanon, New Hampshire); Kevin Stevenson, MD, Logan Mahoney, LPN (Piedmont Orthopaedic Complex, Macon, Georgia); Fabien Bitan, MD, Stephanie Gomez (Manhattan Orthopaedic Spine, New York City, New York); John Stevenson, MD, Ana Marichal (The Orthopaedic Institute, Gainesville, Florida); Donald Sachs, MD, Robin Cambron, MSN, MBA, RN, Missy White, Ana Colburn, RN, Sally Raiden, RN, MSN (Center for Spinal Stenosis and Neurologic Care, Lakeland, Florida); Abhineet Chowdhary, MD, Tina Fortney, RN, BSN (Overlake Hospital Medical Center, Bellevue, Washington); Gowriharan Thaiyananthan, MD, Tungie Williams (BASIC Spine, Orange, California); Michael Oh, MD, Gary Schmidt, MD, Matthew Yeager (Allegheny General Hospital, Pittsburgh, Pennsylvania); David Wiles, MD, Susan Maye, RN, MS (East Tennessee Brain & Spine, Johnson City, Tennessee); Michael Hasz, MD, Carrie Califano (Virginia Spine Institute, Reston, Virginia); William Rosenberg, MD, Pamela McCann, RN, BSN (Mid-west Division-RMC, LLC,-Research Medical Center, Kansas City, Missouri); Jeffrey D. Coe, MD, Julia Coe, Marlene Coe (Silicon Valley Spine, Campbell, California); Jed Vanichkachorn, MD, Jessica Lynch (Tuckahoe Orthopaedics Associates, Richmond, Virginia); Mark C. Gillespy, MD, Sherri Zicker, RN (Orthopaedic Clinic of Daytona Beach, Daytona Beach, Florida); Ralph Rashbaum, MD, Shannon Rusch, BA, CCRC (Texas Back Institute, Plano, Texas); Emily A. Darr, MD, John A. Glaser, MD, Laura Fields, Monica Baczko (Medical University of South Carolina, Charleston, South Carolina).
- Copyright © 2016 ISASS - This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Minimally invasive sacroiliac joint fusion using triangular titanium implants versus nonsurgical management for sacroiliac joint dysfunction: a systematic review and meta-analysis
- Minimally Invasive SI Joint Fusion Procedures for Chronic SI Joint Pain: Systematic Review and Meta-Analysis of Safety and Efficacy
- Patient-Reported and Radiographic Outcomes After Revision Sacroiliac Joint Fusion
- Patient-Reported and Radiographic Outcomes After Revision Sacroiliac Joint Fusion
- Opioid Prescription Monitoring in Preoperative and Postoperative Sacroiliac Joint Fusion Patients
- Minimally Invasive Sacroiliac Joint Fusion vs Conservative Management in Patients With Sacroiliac Joint Dysfunction: A Systematic Review and Meta-Analysis
- Clinical Outcomes Following Minimally Invasive Sacroiliac Joint Fusion With Decortication: The EVoluSIon Clinical Study
- Review of Current Evidence for Minimally Invasive Posterior Sacroiliac Joint Fusion
- Editor's Introduction: Update on Current Sacroiliac Joint Fusion Procedures: Implications for Appropriate Current Procedural Terminology Medical Coding
- Surgical Revision after Sacroiliac Joint Fixation or Fusion