


ABSTRACT
Background: This study compared 7-year safety and efficacy outcomes of activL and ProDisc-L lumbar total disc replacements in patients with symptomatic, single-level lumbar degenerative disc disease (DDD). The objectives are to report 7-year outcomes of the trial, evaluate the outcomes for patients lost to follow-up, and determine whether early outcomes predict long-term outcomes.
Methods: This was a prospective, multicenter, randomized, controlled investigational device exemption study. Eligible patients with symptomatic, single-level lumbar DDD had failed ≥6 months of nonsurgical management. Patients (N = 283) were randomized to receive activL (n = 218) or ProDisc-L (n = 65). At 7 years, data were available from 206 patients (activL, 160; ProDisc-L, 46). Logistic regression models were fit to predict 7-year outcomes for patients lost to follow-up after 2 years.
Results: At 7 years, the activL group was noninferior to the ProDisc-L group on the primary composite endpoint (P = .0369). Both groups showed significant reductions in back/leg pain severity and improvements in disability index and quality-of-life relative to baseline (P < .0001). In both groups, opioid use was significantly reduced at 7 years (0%) relative to baseline (P < .01), and the overall reoperation rates were low (4.6%). activL patients showed a significantly better range of motion (ROM) for flexion-extension rotation than ProDisc-L patients (P = .0334). A significantly higher proportion of activL patients did not report serious adverse events (activL, 62%; ProDisc-L, 43%; P = .011). Predictive modeling indicated that >70% of patients (depending on outcome) lost to follow-up after 2 years would show clinically significant improvement at 7 years if improvements were achieved at 2 years.
Conclusions: The benefits of activL and ProDisc-L are maintained after 7 years, with significant improvements from baseline observed in pain, function, and opioid use. activL is more effective at preserving ROM than ProDisc-L and has a more favorable safety profile. Improvements in other primary and secondary outcomes were similar between both disc designs.
Level of Evidence: 1.
INTRODUCTION
Symptomatic lumbar degenerative disc disease (DDD) is a major cause of back pain, missed work days, and healthcare utilization.1,2 Historically, there has been controversy to support interventional procedures to treat symptomatic DDD. Clinical studies show that when reasonable nonsurgical therapies have been exhausted, lumbar total disc replacement (TDR) is an excellent treatment option in appropriately selected patients.3–13 These scientifically robust, high-level studies report equivalent or improved results for lumbar TDR compared with fusion across various outcomes.3–13 Lumbar TDR has also been shown to be superior to nonsurgical treatment during long-term follow-up.14
Long-term studies have also demonstrated that the likelihood of secondary surgery at adjacent spine segments is higher among patients who undergo fusion than among those who undergo lumbar TDR.4,15–22 A recent study showed no significant difference in adjacent segment degeneration rates at 8 years between patients who underwent lumbar TDR (42%) and those who continued with nonsurgical treatment (40%).23 These data suggest that the progression of adjacent segment degeneration is part of the natural history of DDD but may be accelerated by lumbar fusion. Consistent with clinical evidence, biomechanical data demonstrate that the loss of motion caused by spinal fusion transfers stresses to adjacent spinal regions and causes accelerated degeneration.24,25
The activL US Food and Drug Administration (FDA) investigational device exemption (IDE) randomized study compared a newer generation TDR (ie, activL artificial disc) to an arthroplasty control group (primarily ProDisc-L) instead of fusion.8 The current study represents a longer-term follow-up of the initial IDE trial. The purposes of this study are to (1) report 7-year outcomes of the trial comparing activL and ProDisc-L for the treatment of symptomatic single-level DDD, (2) evaluate the characteristics and outcomes for patient lost to follow-up, and (3) determine whether early outcomes predict long-term outcomes.
METHODS
This prospective, multicenter, randomized controlled IDE trial (NCT00589797)8 was approved by the FDA and institutional review board at each site. In 14 sites, eligible patients reported lumbar pain and back dysfunction due to a radiographically confirmed diagnosis of DDD at a single symptomatic level (L4-L5 or L5-S1) following ≥6 months of nonsurgical management. Appendix 1 describes inclusion and exclusion criteria. Patients provided written informed consent for the study.8
Between January 2007 and December 2009, patients were randomly allocated (2 : 1) to activL or a control disc by permuted block randomization. Sealed envelopes were delivered, which were opened at the time of first incision. Patients were blinded to treatment assignment. Surgeons were blinded to allocation at enrollment, and treatment was assigned at the time of surgery. In patients randomized to the control group, choice of ProDisc-L or Charité was at the investigator's discretion based on preference and experience. Descriptions of the activL, ProDisc-L, and Charité TDRs are previously reported.3,8,26,27 This 7-year analysis focuses on activL and ProDisc-L as Charité is not commercially available. Results, including the full control group, are presented in Appendix 2.
The primary endpoint for this IDE trial was composite treatment success at 2 years, which required patients to meet several criteria involving the Oswestry Disability Index (ODI), neurological status, range of motion (ROM), freedom from secondary surgery, and serious device-related adverse events. Details of the specific criteria for this endpoint are described elsewhere.8 Secondary outcomes included back and leg pain severity on a 0 to 100 visual analog scale (VAS; pain was measured at rest, and patients were considered responders if they achieved ≥20 mm VAS improvement versus baseline; for leg pain severity, no worsening in the other leg was permitted), ODI, quality-of-life assessed with the 36-item short form survey (SF-36), patient satisfaction, return to work, opioid usage, ROM, radiographic evaluations of device status, adverse events, and reoperations (defined as any surgical procedure at the level of the original implant that does not include removal, modification, or addition of any components of the system). A serious adverse event (SAE) was defined as any event that was fatal, life-threatening, required prolonged hospitalization, resulted in permanent anatomic or physiological impairment, caused a malignant tumor, or resulted in distress, congenital anomaly, or death of a fetus. Serious device-related adverse events were those SAEs attributable to TDR; new back or leg pain and/or worsening of existing back or leg pain fell under this category irrespective of whether or not the event involved hospitalization.
Patients returned for follow-up visits at 6 weeks, 3 months, 6 months, and annually thereafter for 7 years. A physical examination, neurological assessment, and 6-view x-rays were performed at follow-up visits.
Additional study design details are reported elsewhere.8
Statistical Methods
Continuous data are reported as mean ± standard deviation or median (min-max) depending on normality assumptions. Categorical data are reported as frequencies and percentages. The Farrington-Manning noninferior test with a margin of 10% tested for noninferiority between the activL and ProDisc-L groups in the primary endpoint. Comparisons of continuous data were made using a two-sample t test, while comparisons of categorical data were performed using the Fisher exact testing. Time to event data were analyzed using Kaplan-Meier methods with a log-rank test for group differences. Statistical significance was set at P < .05. Multiple imputation accounted for missing data.
The relationship between results at 7 years and previous scheduled visits was assessed to understand if short-term results predict long-term results. Only patients with complete data through 7 years were analyzed in logistic regression analyses; therefore, complete case methods, rather than multiple imputation, were used here. Outcomes were analyzed dichotomously as “clinically significant improvement” (CSI) relative to baseline, where CSI was defined as maintenance or improvement for radiographic measures, ≥15 points reduction for function (ODI), ≥20 mm reduction for pain, and ≥15% increase for quality of life. Odds ratios (ORs) informed the association between CSI status at 2 and 7 years. The McFadden pseudo-R2 and Hosmer-Lemeshow test assessed goodness of fit and predictive power of logistic regression models,28 which were used to predict 7-year outcomes for patients lost to follow-up after 2 years. Outcomes were also analyzed as continuous values using correlation coefficients between different study time points.
RESULTS
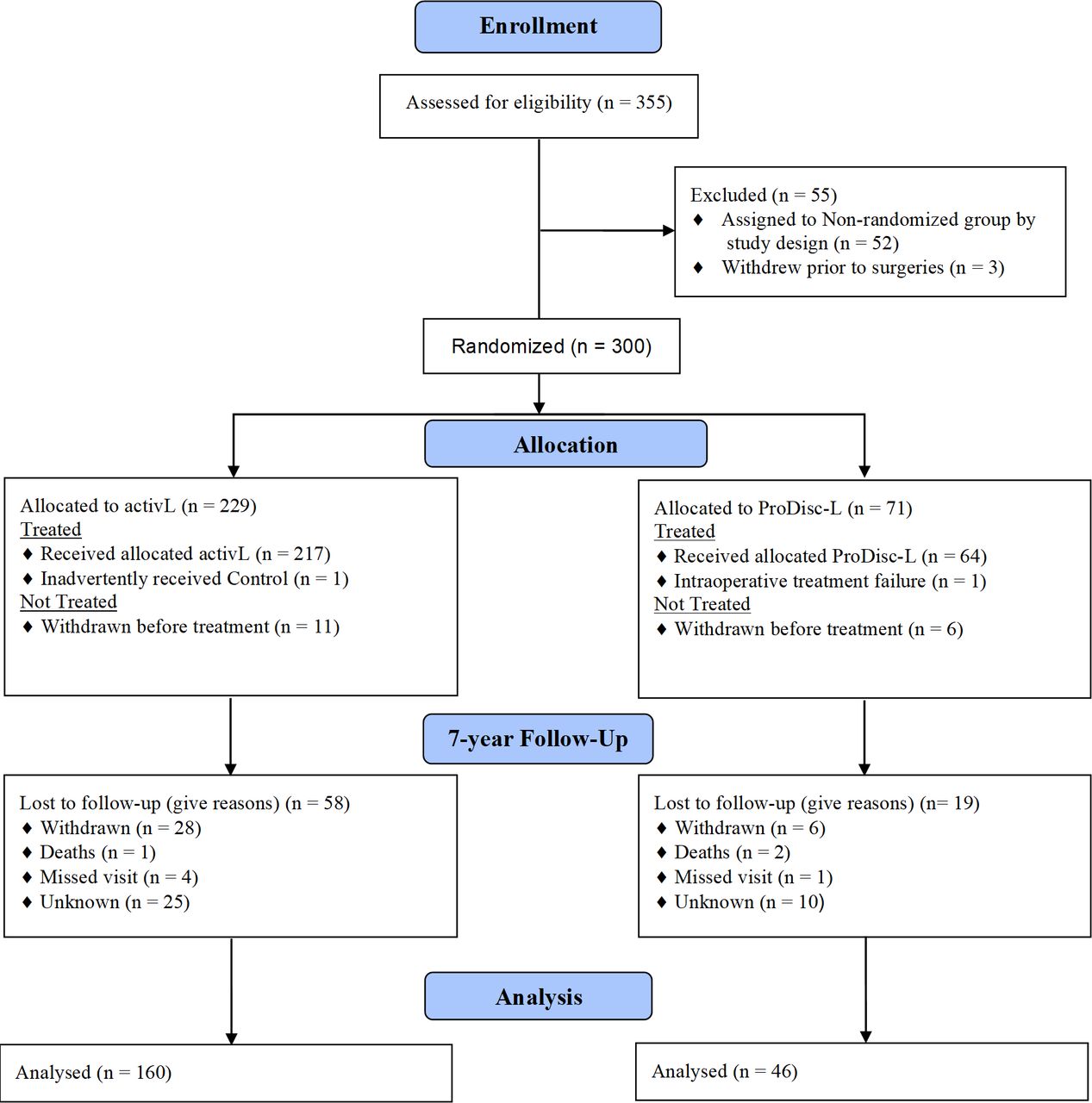
Between January 2007 and December 2009, 283 patients were treated with the activL (n = 218) or ProDisc-L (n = 65). Approximately 73% (206/283) of patients returned for the 7-year follow-up visit (Figure 1). Charité-treated patients are not discussed here but are presented in appendices as part of the full control group (Appendices 1 and 3).

CONSORT flow diagram, activL versus ProDisc-L.
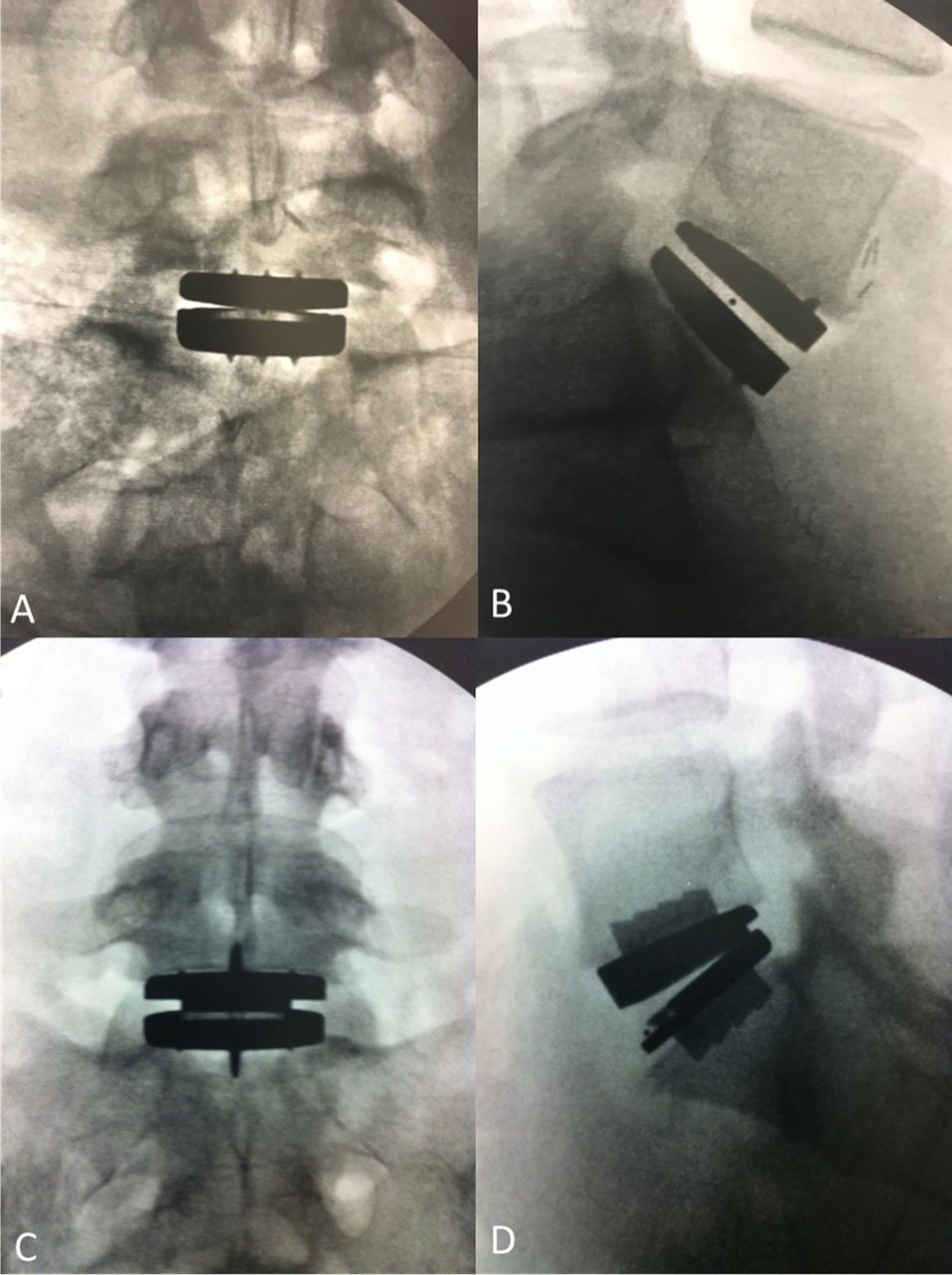
Baseline variables were generally well matched between groups, with the exception of hepatic/biliary conditions (Table 1). Mean age at time of surgery was 40 years, and 49.5% of patients were female. The total number of patients with L5-S1 or L4-L5 disease was 150 and 56, for the activL or ProDisc-L groups, respectively. Baseline characteristics were also similar between patients that remained at 7 years and those lost to follow-up except for SF-36, instability, and lateral rotation (Table 2). See radiographs of TDR implants (Figure 2).
Baseline characteristics for patients at randomization and patients remaining at 7 years.
Baseline characteristics for patients remaining at 7 years and those lost to follow-up.
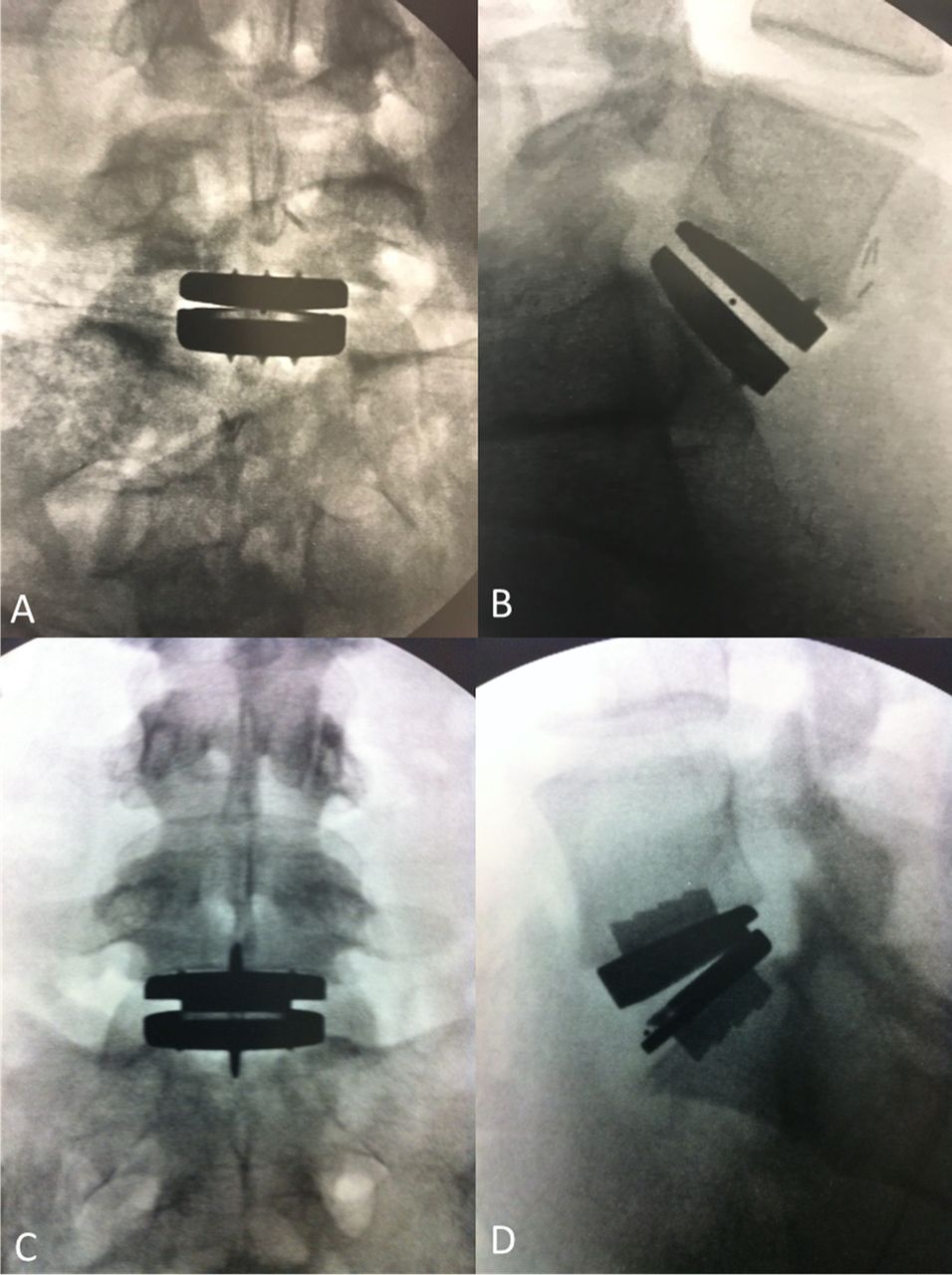
Radiographs of total disc replacement implants. (A) Anteroposterior view of activL, (B) lateral view of activL, (C) anteroposterior view of ProDisc-L, and (D) lateral view of ProDisc-L.
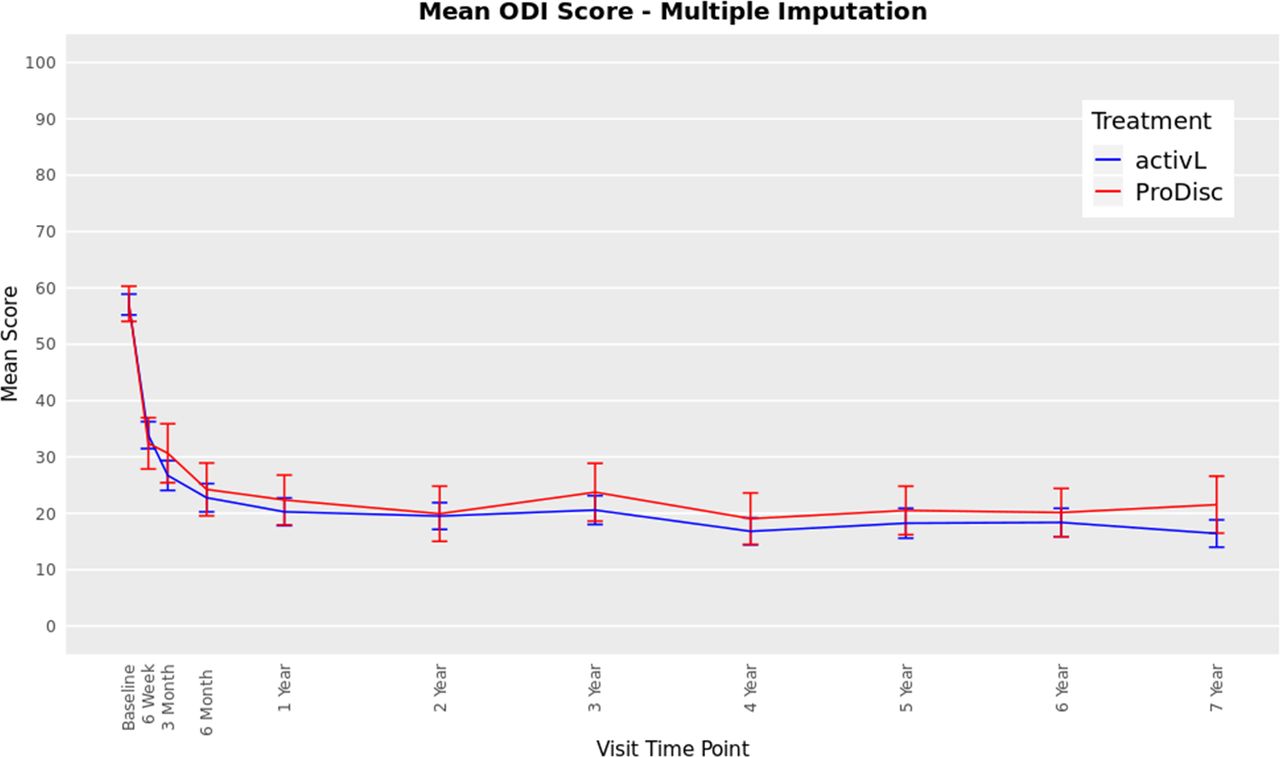
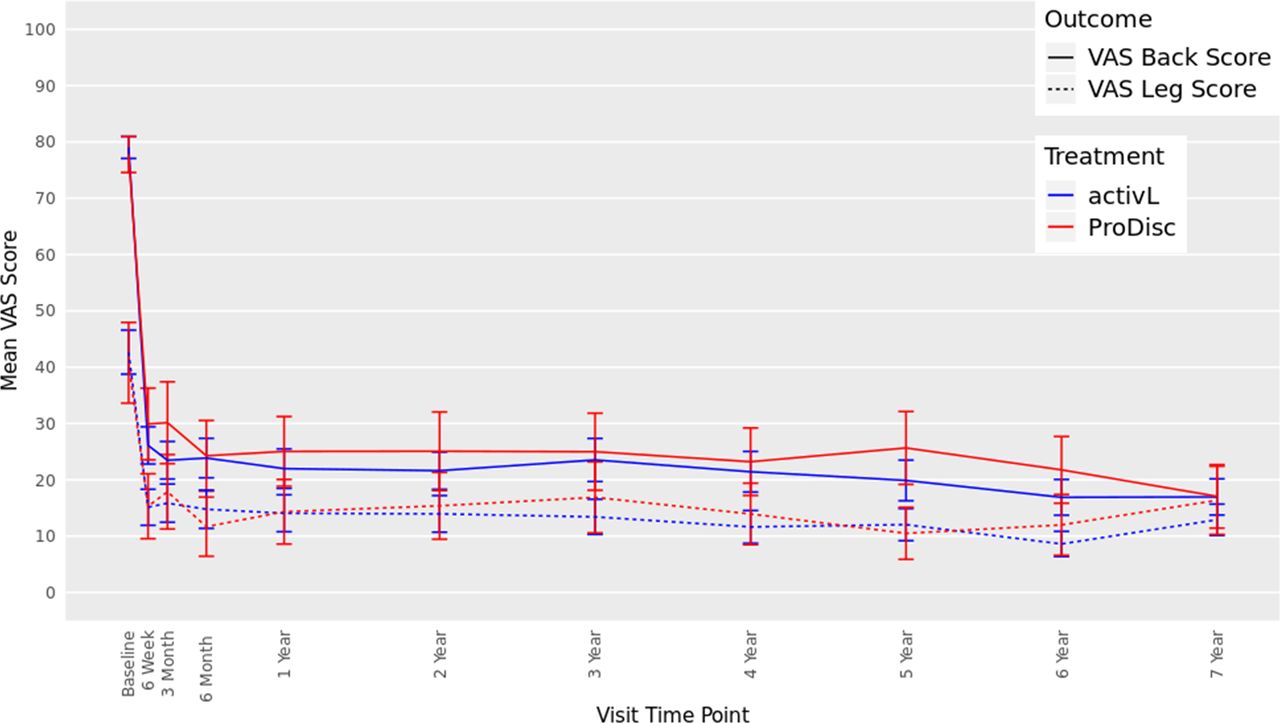
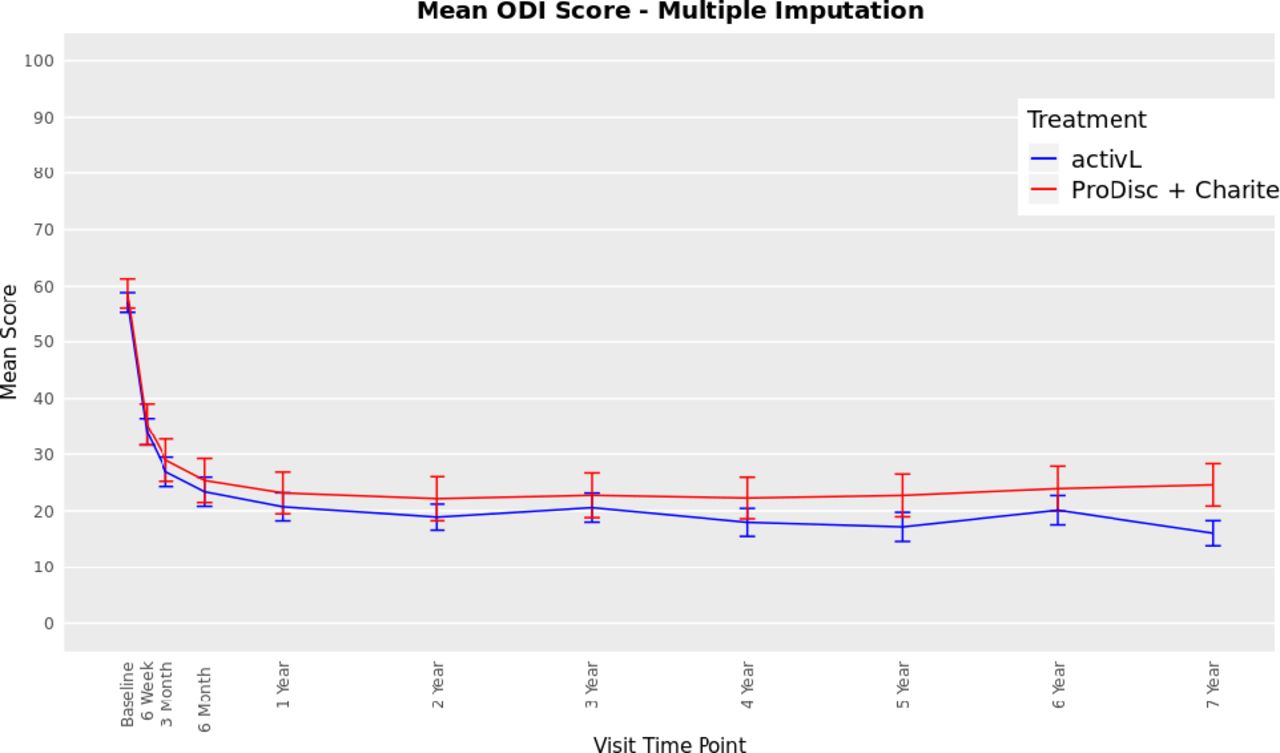
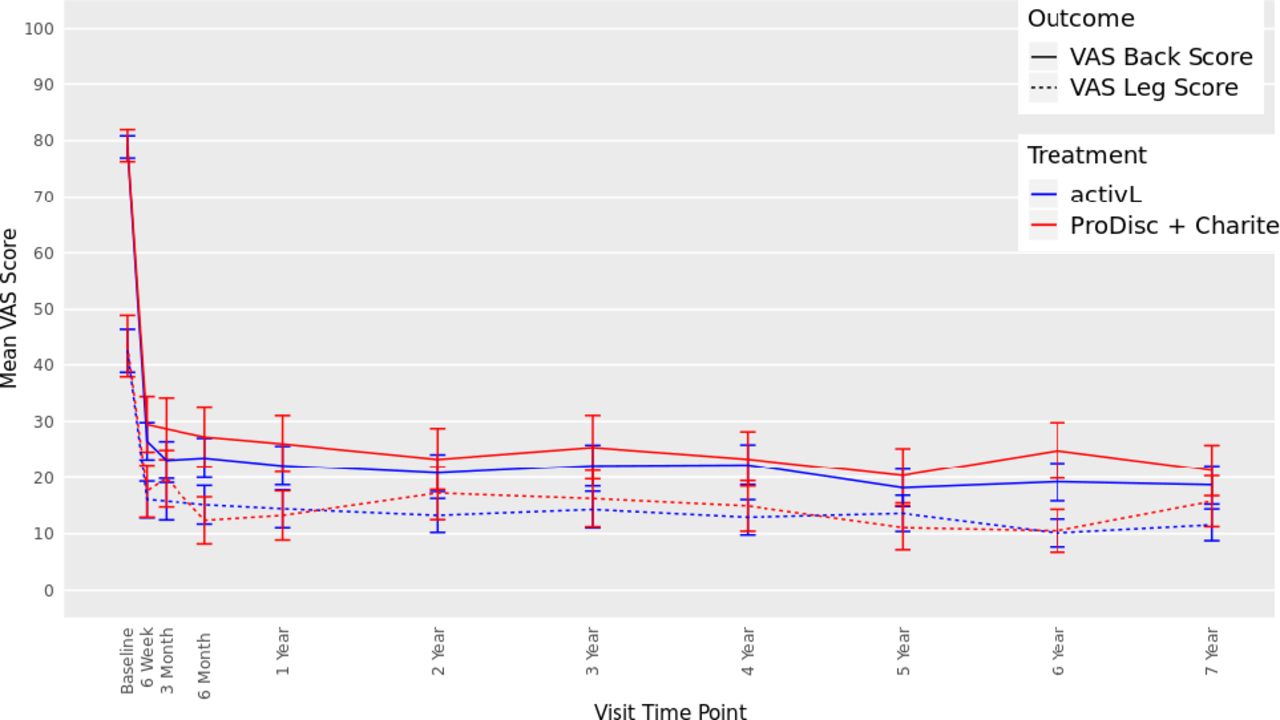
The ODI scores decreased from 57 at baseline to 16 at 7 years with the activL patients and from 59 to 22 with ProDisc-L patients (P < .0001 for change from baseline) (Figure 3). For the activL patients, mean VAS back and leg pain scores decreased from 79 mm to 17 mm and from 43 mm to 13 mm, respectively (P < .0001 for change from baseline) (Figure 4). Similar results were observed for ProDisc-L patients (VAS back score, 78 mm to 17 mm; leg score, 41 mm to 16 mm; change from baseline, P < .0001). There were no statistically significant differences between the activL and ProDisc-L at any time point in ODI or VAS.
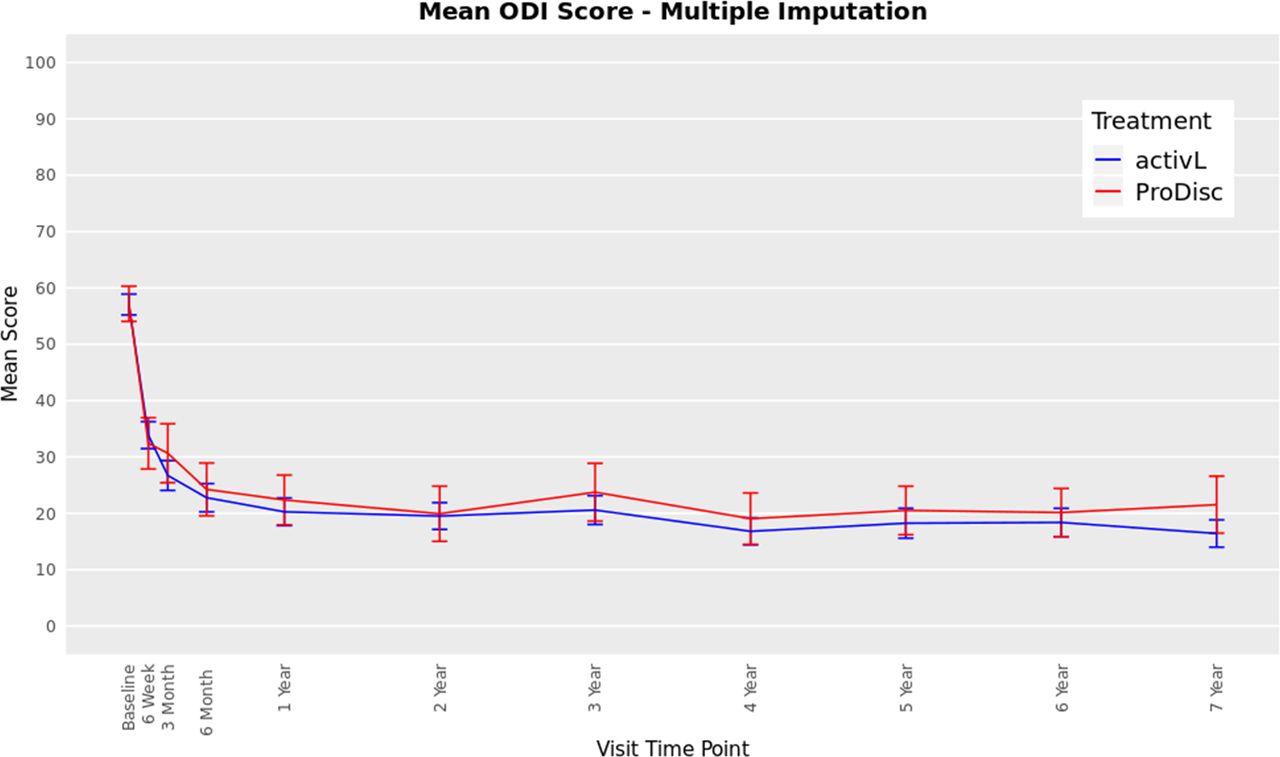
Oswestry Disability Index (ODI) through 7 years after treatment for activL versus ProDisc-L artificial discs. Multiple imputation was used for missing patient data. Values are presented as mean ± 95% confidence interval.

Visual analog scale (VAS) back and leg score through 7 years after treatment for activL versus ProDisc-L artificial discs. Multiple imputation was used for missing patient data. Values are presented as mean ± 95% confidence interval.
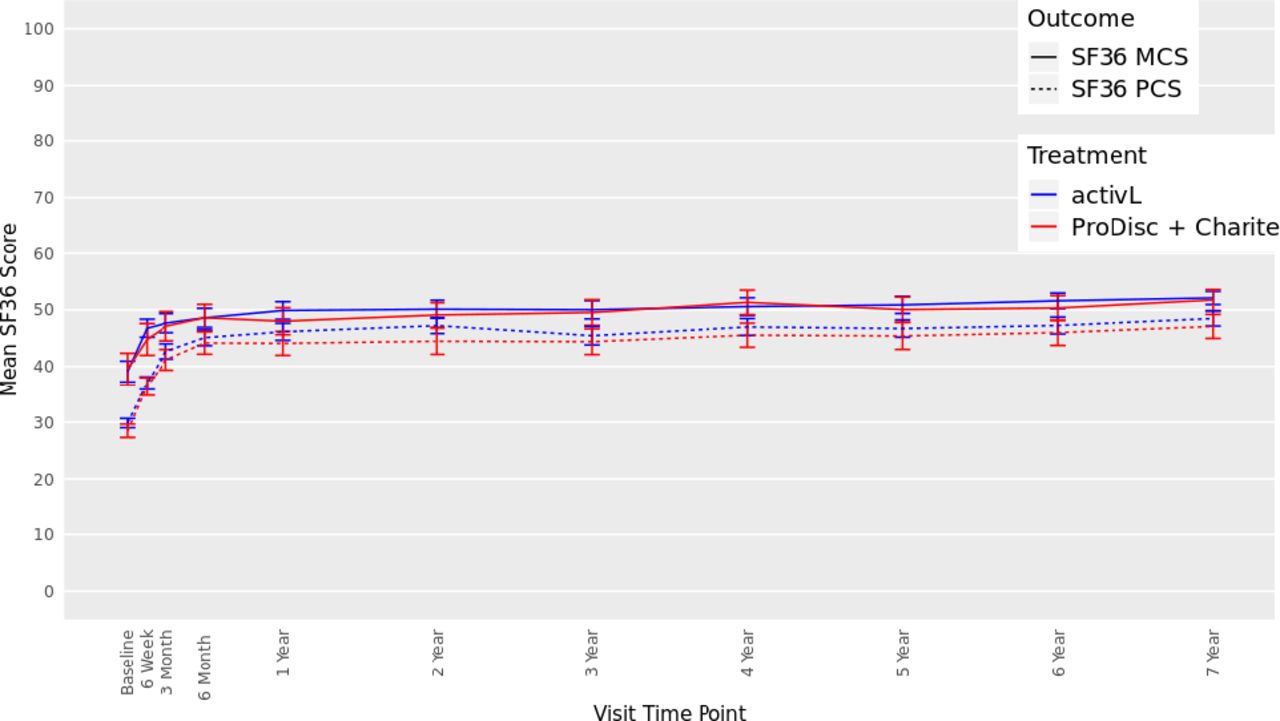
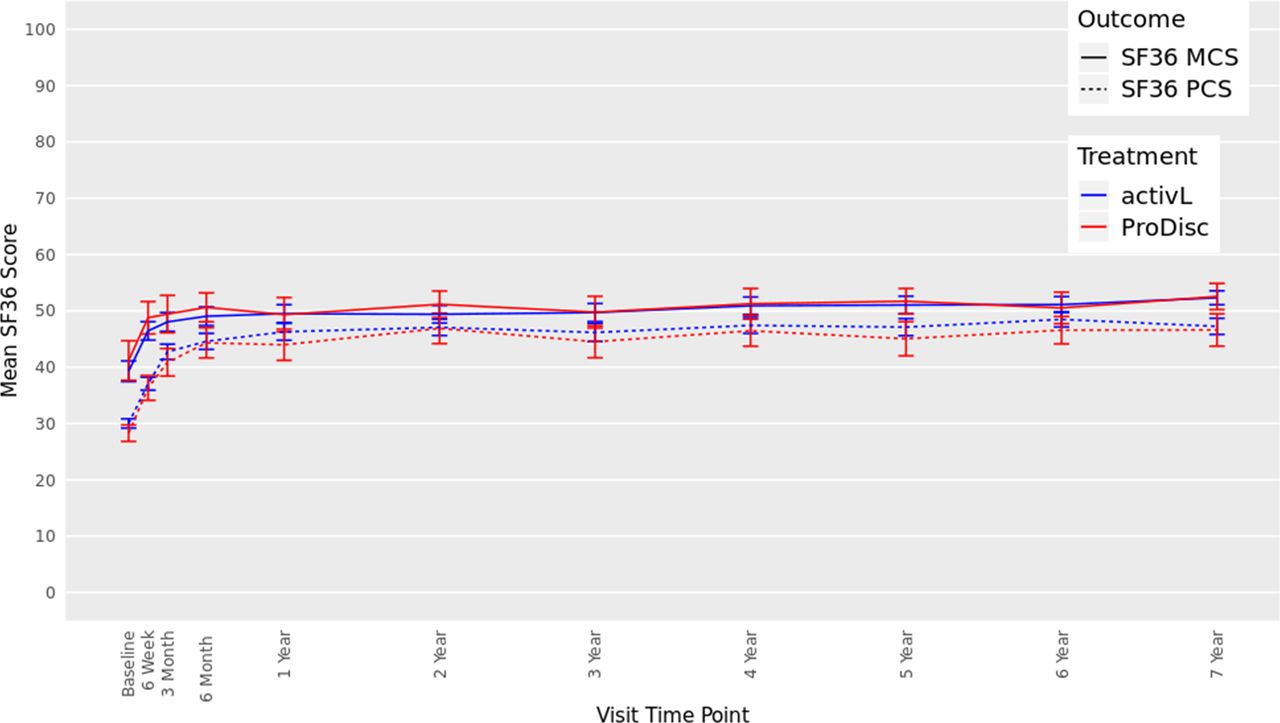
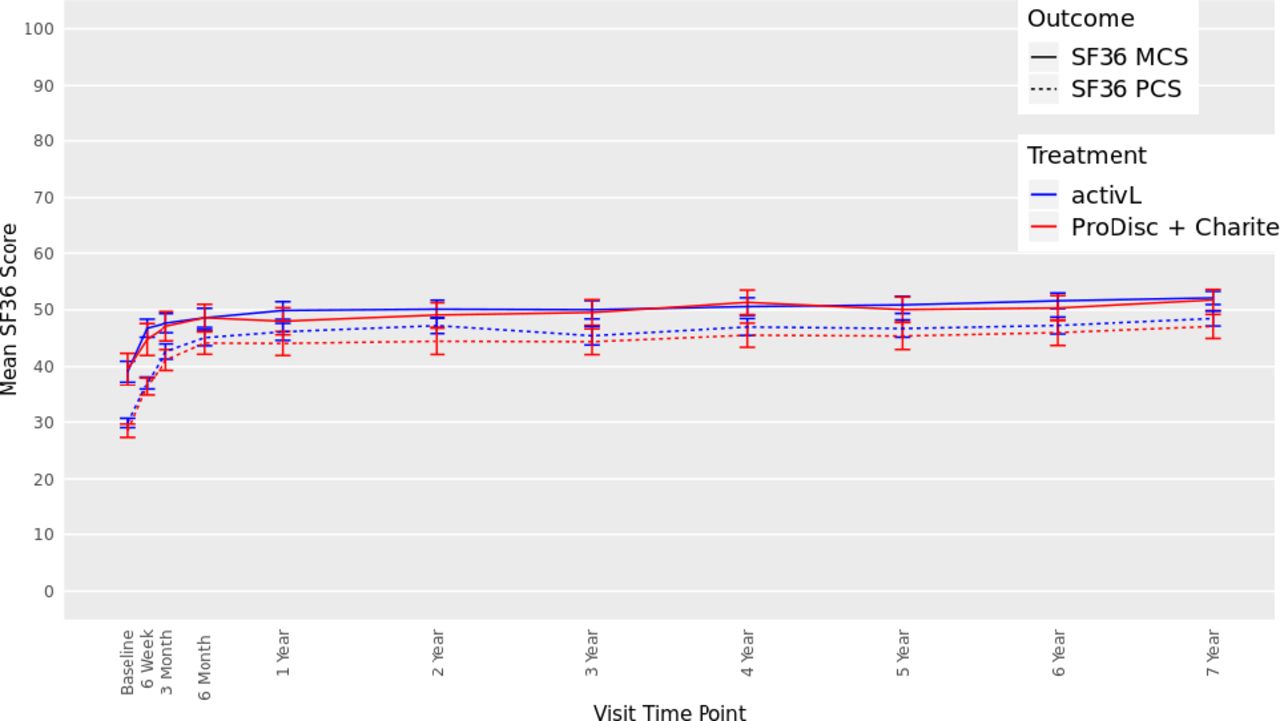
There were also significant improvements from baseline in SF-36 scores in both groups at 7 years (Figure 5). The mean Physical Component Summary improved by 17.2 points and 18.3 points, for the activL and ProDisc-L patients, respectively (P < .0001). Moreover, the mean Mental Component Summary also significantly improved compared with baseline (P < .0001) (activL, 13.1 points; ProDisc-L, 11.4 points). There were no statistically significant differences between activL and ProDisc-L.

Thirty-six-item short form survey (SF-36), Mental Component Summary (MCS), and Physical Component Summary (PCS) data through 7 years after treatment for activL versus ProDisc-L artificial discs. Multiple imputation was used for missing patient data. Values are presented as mean ± 95% confidence interval.
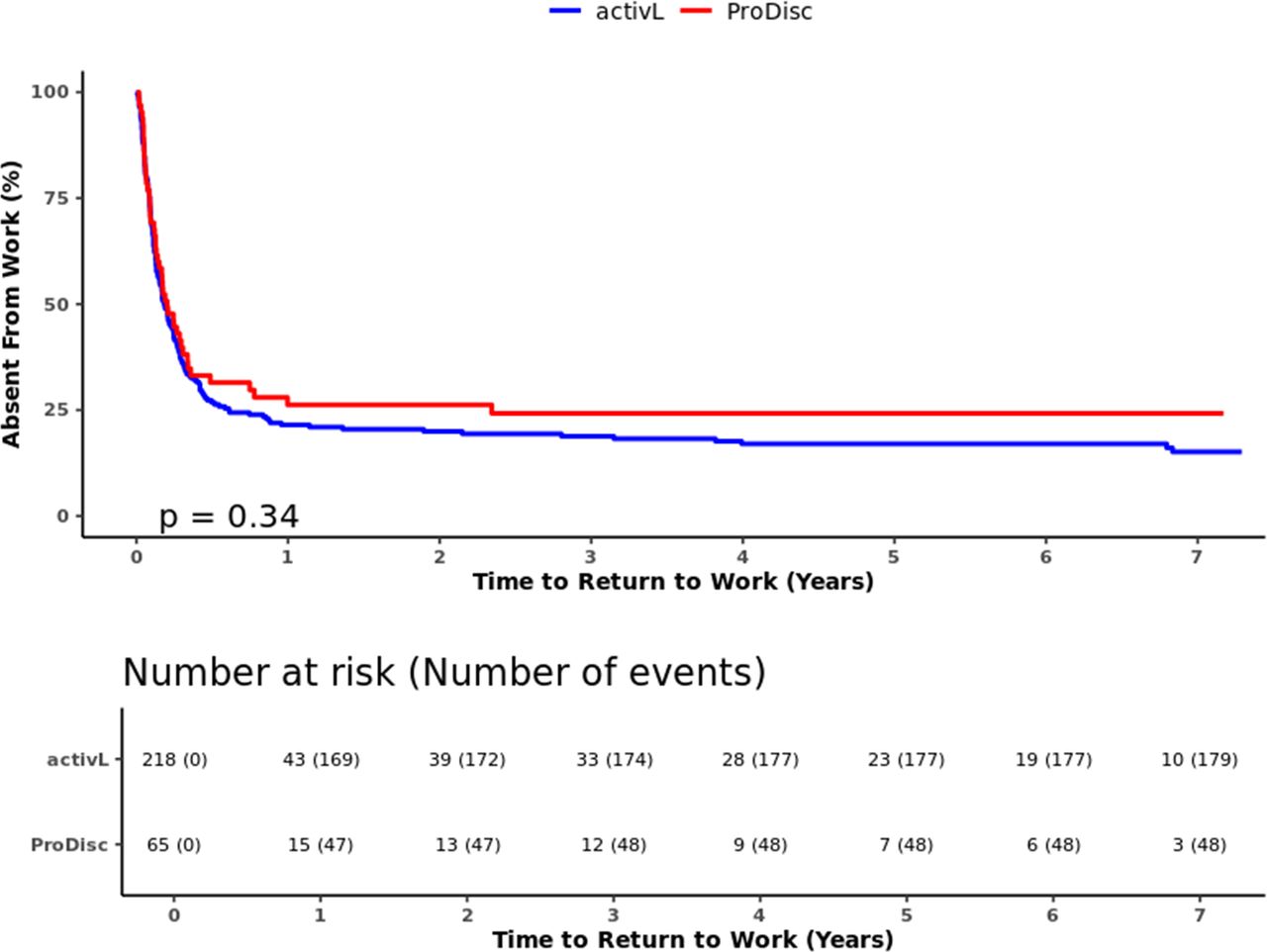
Overall, there was a trend toward a higher proportion of the activL patients willing to have surgery again (activL, 97.2%; ProDisc-L, 91.9%; P = .0749) and having felt that treatment was effective in eliminating symptoms than ProDisc-L patients (activL, 90.6%; ProDisc-L, 82.3%; P = .1072). Also, a higher proportion of the activL than ProDisc-L patients reported being satisfied with treatment, but results were not statistically significant (activL, 95.3%; ProDisc-L, 93.5%; P = .526). The activL patients also had a reduced median return to work time (67 days (activL) versus 74 days (ProDisc-L); log-rank = 0.34); however, these results were also not statistically significant (Figure 6).

Time to return to work through 7 years for activL versus ProDisc-L artificial discs. Kaplan-Meier estimate is 82% with activL and 74% with ProDisc-L. Log-rank P value = .34.
At 7-year follow-up, the composite treatment success rate was not significantly different between groups (activL, 30.7%; ProDisc-L, 29.2%; P = .8787). The activL was significantly noninferior compared with the ProDisc-L group (P = .0369).
The activL patients were associated with significantly greater flexion-extension rotation than ProDisc-L patients after 7 years (Table 3), and radiographic variables were not significantly different between groups at baseline (Table 1). At 7 years, 0% of the activL patients were classified as having clinically significant class IV heterotopic ossification; this was numerically lower than the proportion observed with ProDisc-L (3.1% [2/65], P = .0521).
Radiographic findings at 7 years for activL versus ProDisc-L artificial discs.a
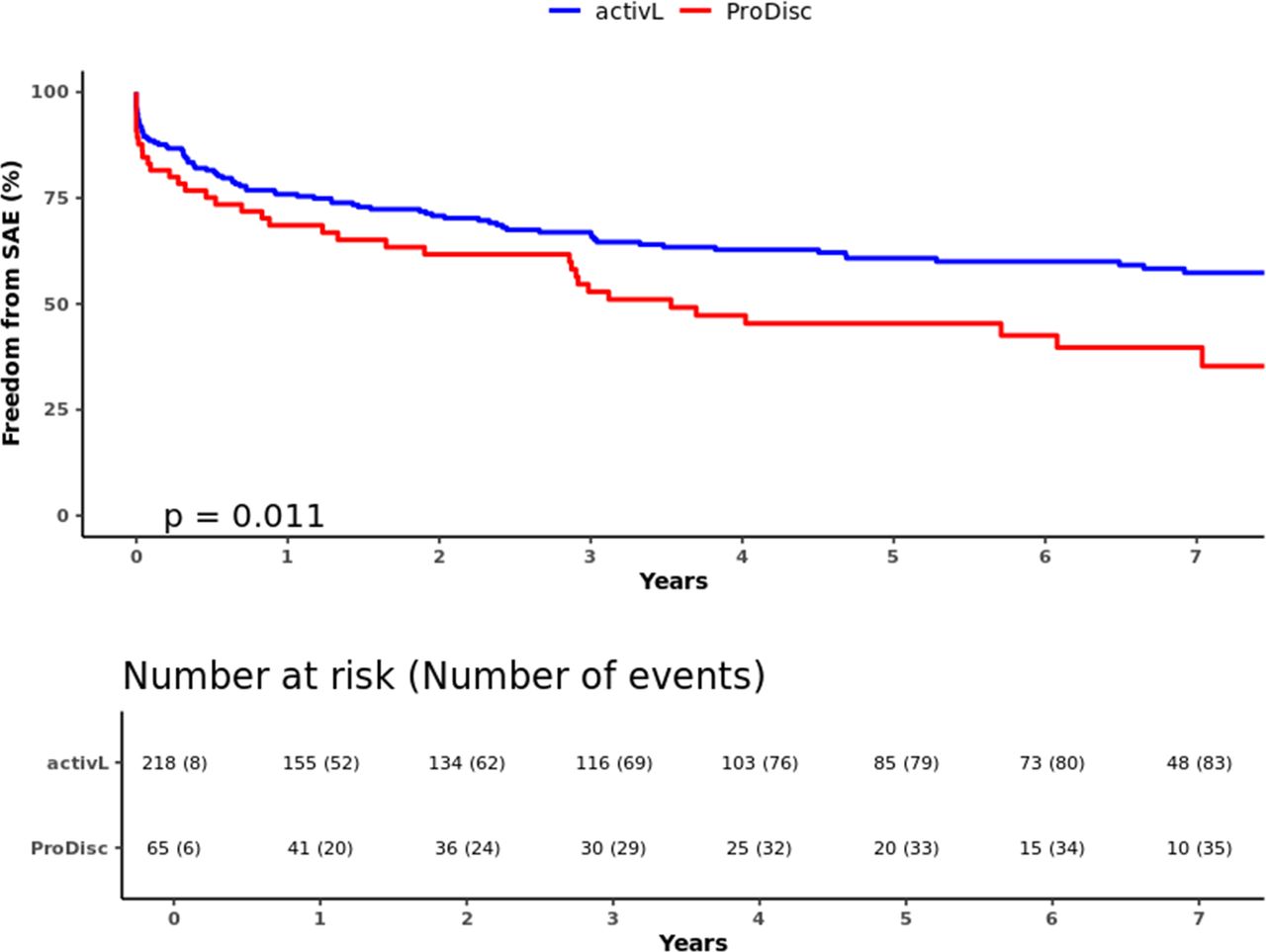
In total, there were 227 SAEs reported through 7 years (activL, 163; ProDisc-L, 64) (Table 4). Of the 227 SAEs, 17% (39/227) were classified as device related, and the most common type of device-related SAE was pain in the lumbar spine and/or lower extremities (24/39, 61.5%). The activL patients had a significantly lower risk of SAEs than ProDisc-L patients through 7 years; freedom from SAEs was 61.5% with activL and 43.1% with ProDisc-L (P = .011) (Figure 7). The trial also demonstrated a very low reoperation rate, as over 95% (270/283) of the activL and ProDisc-L patients were reoperation-free through 7 years (P > .05 between groups).
Serious adverse events through 7 years for activL versus ProDisc-L artificial discs.
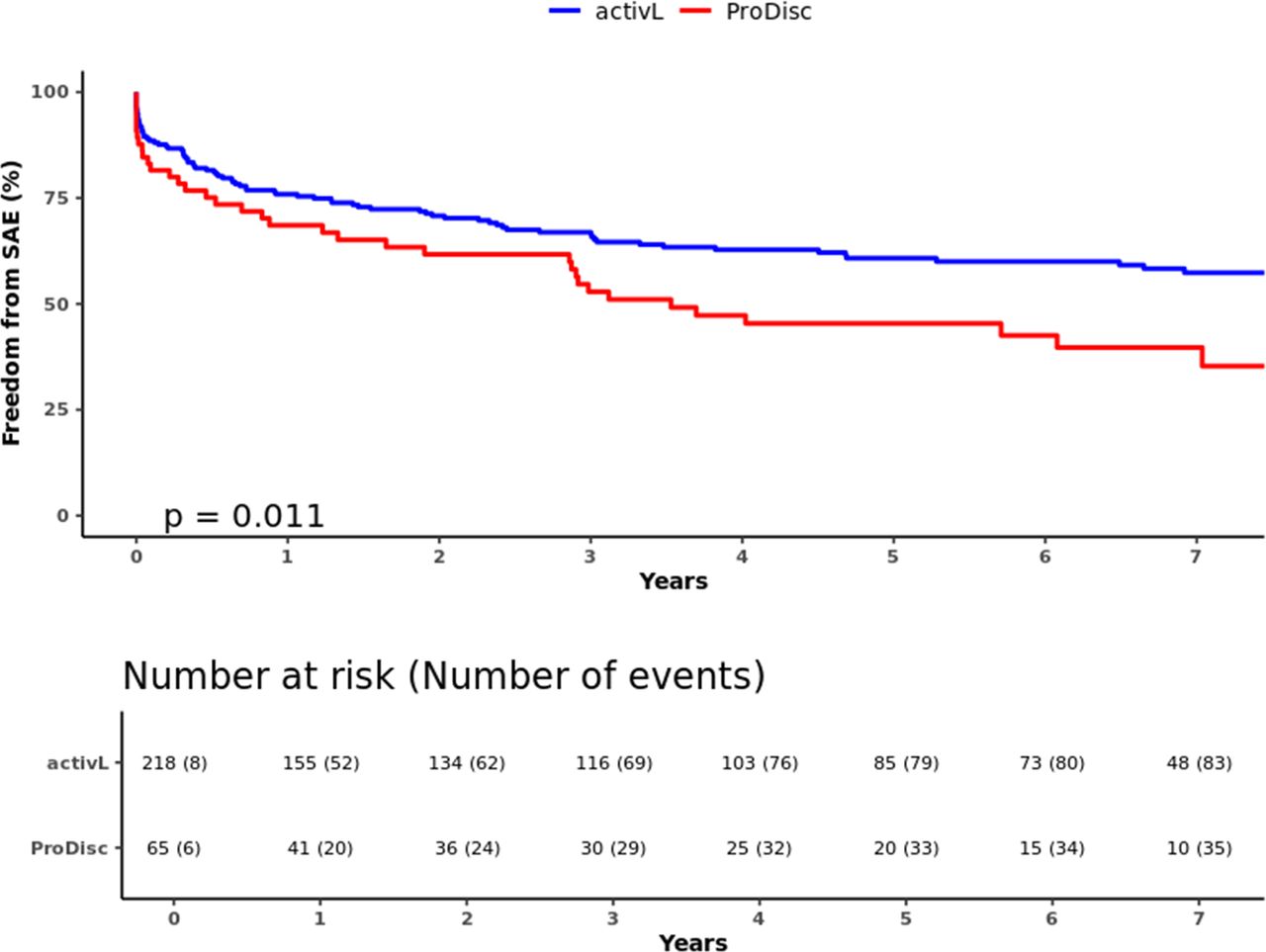
Freedom from serious adverse events through 7 years for activL versus ProDisc-L artificial discs. Kaplan-Meier estimate is 61.5% with activL and 43.1% with ProDisc-L. Log-rank P value = .011.
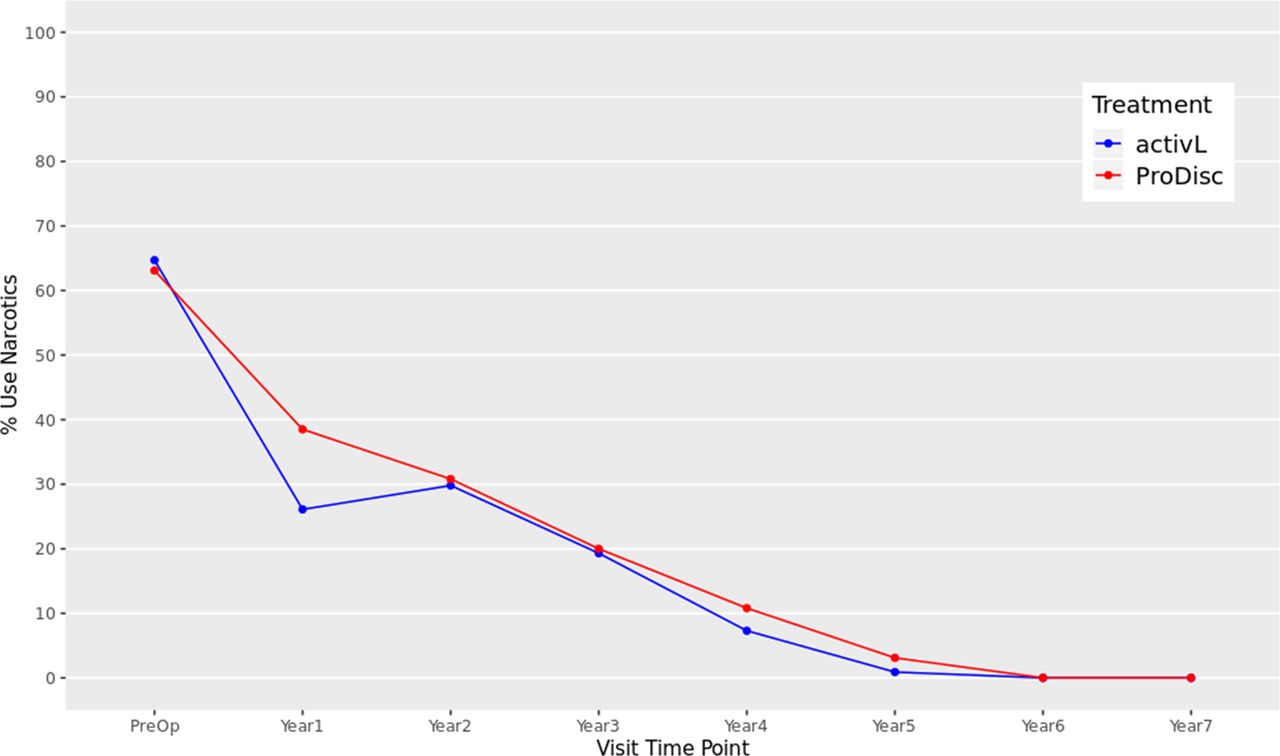
At baseline, 64.7% of the activL and 63.1% of ProDisc-L patients were using opioids. Utilization was significantly reduced at 1 through 7 years for both the activL and ProDisc-L patients compared with baseline (P < .01), and there were no patients using opioids after 6 or 7 years (Figure 8).
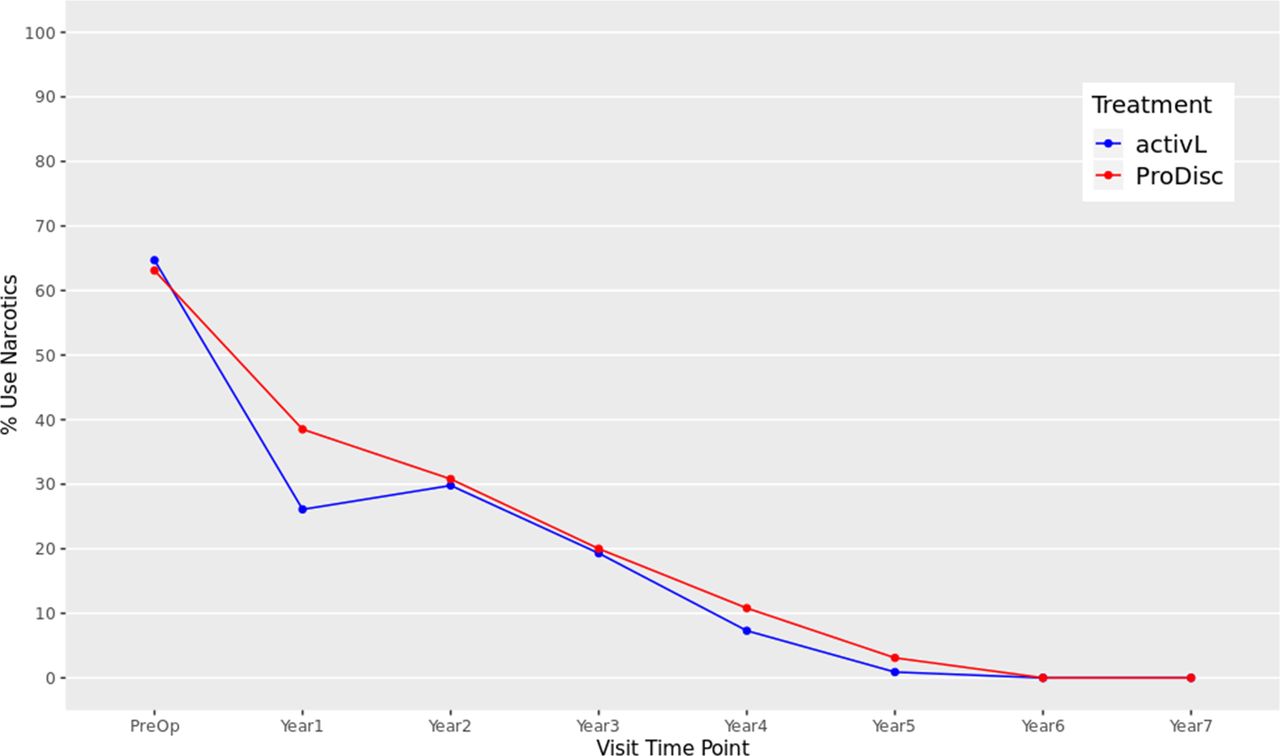
Opioid usage through 7 years after treatment for activL versus ProDisc-L artificial discs. At each follow-up time point postoperation, a significantly lower proportion of patients with activL and ProDisc-L total disc replacement (TDR) devices used opioids than at baseline (P < .01 at each time point). Multiple imputation was used for missing patient data.
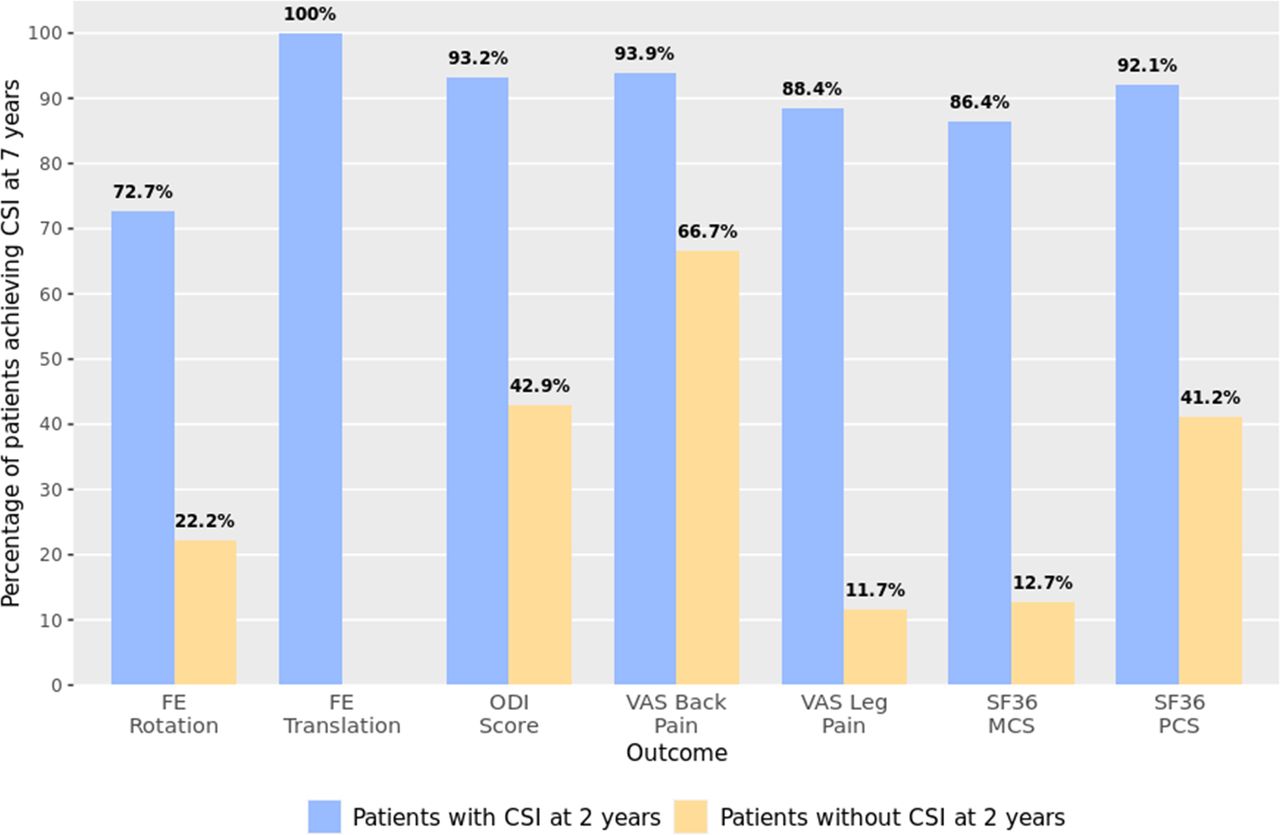
Patients were more likely to experience improvements at 7 years if they experienced improvement at earlier visits. The percentage of patients achieving CSI at 7 years was significantly higher for patients that had CSI at 2 years for pain, function, quality of life, and radiographic outcomes. For ODI, 93% (124/133) of patients with CSI at 2 years also had CSI at 7 years, while only 43% (6/14) of patients without CSI at 2 years achieved CSI at 7 years; the estimated OR of 18.37 (95% confidence interval [CI], 5.25 to 64.52) indicated a strong association between 2- and 7-year CSI. Similar results were observed for other outcomes, such as flexion-extension rotation (OR, 9; 95% CI, 4 to 21), VAS leg score (OR, 58; 95% CI, 21 to 161), and SF-36 mental component summary (OR, 44; 95% CI, 17 to 116) (Table 5 and Figure 9).
Contingency table from early versus late outcomes predictors analysis for activL versus ProDisc-L artificial discs.a

Percentage of patients who achieved clinically significant improvement (CSI) at 7 years, stratified by their CSI status from predictor analysis for activL versus ProDisc-L artificial discs. Complete case analysis conducted (no imputations).
As baseline characteristics of patients who remained at 7 years were similar to those lost to follow-up, the logistic regression analyses were used to predict long-term outcomes in those who dropped out. Of the 27% of patients lost to follow-up, 51% to 100% of these patients achieved CSI at 2 years, depending on outcome. Regression analyses predicted that most of these patients (>70% across outcomes) would have achieved CSI at 7 years. For instance, 72.7% (42/58) of patients lost to follow-up were expected to have achieved CSI for flexion-extension rotation at 7 years, 93.2% (70/75) for ODI score, and 88.4% (42/47) for VAS leg pain score.
Nearly all logistic regression models fitted had reasonably high McFadden pseudo-R2 values. Depending on the outcome, the R2 values ranged from .17 to .47, indicating that the models had strong predictive power. The Hosmer-Lemeshow test showed no evidence of poor fit, as the P values were greater than .05 (Table 5).
Similar results were found with analysis of continuous outcomes. Correlations between 7-year outcomes and previously scheduled visits were significantly positive, and magnitude of correlations increased as the trial progressed.
DISCUSSION
The activL artificial disc was approved by the FDA in 2015 based on positive 2-year outcomes, with a high follow-up rate of 83% and 80% for the activL and control groups, respectively. Peer-reviewed reports of both 2- and 5-year outcomes have been published, which show maintenance of significant improvements from baseline for many outcomes.8,9
The long-term follow-up results of this IDE trial demonstrated that statistically significantly improved VAS and ODI, observed within weeks of surgical intervention, were maintained to 7 years. Reoperation rates for both activL and ProDisc-L patients remained low. Importantly, there was no observed increase in SAEs between years 5 and 7, and the great majority of SAEs were not device related. Freedom from SAEs was observed to be higher with activL than with ProDisc-L, primarily due to a lower number of leg and back pain device-related SAEs. This result correlates with efficacy outcomes, where the VAS was also observed to be numerically (but not statistically) lower for activL patients throughout the study. Given that VAS scores were collected annually and SAEs were collected at any time, it makes clinical sense that these 2 outcomes are not entirely correlated. For example, after reporting SAEs, patients may take pain medication, which can impact VAS score at the following scheduled visit.
A very important finding from this study was that opioid use in this chronic low back pain population was reduced to 0% after 7 years from a preoperative rate of 65%. To help interpret these findings, the impacts of recent opioid preventative measures are worthy of consideration. The patients in this study were randomized between 2007 and 2009, with follow-up occurring over the past decade, which is similar timing to the latest phase of the opioid crisis and its peak in morbidity and mortality.29–31 A study that evaluated the average opioid prescription rate between 2009 and 2010 reported that 58.9% of patients diagnosed with back pain had been prescribed opioids, which suggests that opioid use was higher in our trial population at study initiation than the average population with backpain.32 Further, three observational studies that evaluated opioid use in patients that have undergone lumbar fusion for DDD during similar timeframes as our trial enrollment period, with follow-up periods between 1 and 6 years, reported the rate of postoperative opioid use ranging from 27% to 57%.33–35 Given these literature findings, as well as the positive results in pain outcome measures from our trial, we conclude that the reduction in opioid use in our study can be reasonably attributed to the lumbar TDR surgical intervention.
Clinically, both activL and ProDisc-L showed similar results for several efficacy outcomes. Radiographically, the activL continued to show statistically significant improvements compared with ProDisc-L in flexion-extension rotation. Better ROM using activL may be explained by the ability of the polyethylene core to translate 2 mm in flexion-extension, thus self-adjusting the implant's center of rotation. Another possible explanation is the availability in the activL of an 8.5 mm height. Heterotopic ossification (class IV) occurred less frequently with the activL with borderline statistical significance, possibly also contributing to improved ROM. More natural motion and proper sizing are intended to reduce mechanical stress at both the facet joints and on the adjacent levels.36
Our analysis shows that TDR patients were more likely to experience improvements across outcomes at 7 years if they had improvements at earlier timepoints. Predictive analytics also demonstrated that patients who had CSI at 2 years, but were lost to follow-up thereafter, were highly likely to maintain such improvements at 7 years. We conclude that had follow-up rates been higher in this study, we would expect similar efficacy and safety outcomes to those reported with the current analysis. Short-term data can be a predictor of later outcomes, allaying concerns that there will be a progressive increase in the need for additional medical and surgical care services for patients lost to follow-up.
Ultimately, this study reported a relatively high follow-up rate of 73% after 7 years, where loss to follow-up was defined as centers having lost contact with patients. It is noteworthy to mention that follow-up rates calculated according to FDA definitions are slightly different in that loss to follow-up includes patients who had died of unrelated causes or had withdrawn earlier in the study. Another key strength was the use of two separate statistical methods to address patients lost to follow-up. First, multiple imputation techniques were used as a rigorous method to increase the sample size for outcomes assessment and handle missing data. Second, logistic regression models with high goodness-to-fit results assessed the association between early and late outcomes for patients that had data available and were then used to predict long-term results for those lost to follow-up after 2 years. The combination of such statistical methods gives us high confidence in our long-term results.
A key limitation of this study is the use of an IDE population with strict inclusion criteria. As inclusion criteria are broadened in general practice, some outcome measures may not maintain consistency in level of improvements. Conversely, the IDE criteria have clearly identified patients who should do well both clinically and radiographically after implantation for up to 7 years.
CONCLUSION
Seven-year outcomes of a multicenter, randomized trial support TDR as a safe and effective treatment for single-level lumbar DDD in patients who have failed to improve with nonoperative care. Patients show an early and significant improvement in pain and impairment following recovery from surgery, and statistically significant improvements are maintained through 7-year follow-up. activL was more effective at preserving motion and had a more favorable safety profile than ProDisc-L, although other efficacy outcomes were similar between discs. Encouragingly, opioid medication use was 0% at 7-year follow-up in these patients.
ACKNOWLEDGMENTS
The authors thank Chris Cameron of CRG-EVERSANA for statistical expertise and Katie Kleinschuster, Tabitha Pitten, and Andrea Vovk of Aesculap (Center Valley, PA) for constructive discussions on study design.
APPENDIX 1. ACTIVL INVESTIGATIONAL DEVICE EXEMPTION RANDOMIZED TRIAL INCLUSION AND EXCLUSION CRITERIA
Key inclusion criteria
Skeletally mature adults aged 18 to 60 years.
Radiographic evidence of lumbar degenerative disc disease based on identification of any of the following characteristics by magnetic resonance imaging scan:
○ Instability (≥3 mm translation or ≥5° angulation).
○ Osteophyte formation of facet joints or vertebral endplates.
○ Scarring/thickening of ligamentum flavum, annulus fibrosis, or facet joint capsule.
○ Herniated nucleus pulposus.
○ Facet joint degeneration.
○ Vacuum phenomenon.
Single-level symptomatic disease at L4/L5 or L5/S1.
Minimum of 6 months of unsuccessful conservative treatment.
Minimum Oswestry Disability Index score of 40/100.
Minimum visual analog scale back pain score of 40/100 mm.
Surgical candidate for an anterior approach to the lumbar spine.
Key exclusion criteria
Previous surgery at any lumbar level, other than intradiscal electrothermal therapy, percutaneous nucleoplasty, microdiscectomy, hemilaminectomy, or laminotomy.
Chronic radiculopathy, defined as unremitting pain with a predominance of leg pain symptoms greater than back pain symptoms extending over a period of at least 1 year.
Anatomically unsuitable for total disc replacement based on preoperative radiographic assessment.
Myelopathy.
Previous compression or burst fracture at index level.
Sequestered herniated nucleus pulposus with migration.
Midsagittal stenosis <8mm (by magnetic resonance imaging).
Spondylolysis.
Lumbar scoliosis (>11° sagittal plane deformity).
Spinal tumor.
Active systemic infection or infection at the site of surgery.
Facet ankylosis or severe facet degeneration.
Continuing steroid use or prior use for more than 2 months.
Pregnancy or planning to become pregnant within the next 2 years.
Morbid obesity (body mass index of >35).
Osteoporosis, osteopenia, or metabolic bone disease.
History of rheumatoid arthritis, lupus, or other autoimmune disorder.
Ankylosing spondylitis.
Abdominal pathology that would preclude the abdominal surgical approach.
APPENDIX 2. RESULTS OF ACTIVL VERSUS CONTROL (PRODISC-L + CHARITÉ)
Radiographic findings at 7 years for activL versus control.a
Serious adverse events through 7 years for activL versus control (ProDisc-L + Charité).

Oswestry Disability Index (ODI) through 7 years after treatment for activL versus control (ProDisc-L + Charité). Multiple imputation was used for missing patient data. Values are presented as mean ± 95% confidence interval.

Visual analog scale (VAS) back and leg score through 7 years after treatment for activL versus control (ProDisc-L + Charité). Multiple imputation was used for missing patient data. Values are presented as mean ± 95% confidence interval.
Thirty-six-item short form survey (SF-36), mental component summary (MCS), and physical component summary (PCS) through 7 years after treatment for activL versus control (ProDisc-L + Charité). Multiple imputation was used for missing patient data. Values are presented as mean ± 95% confidence interval.

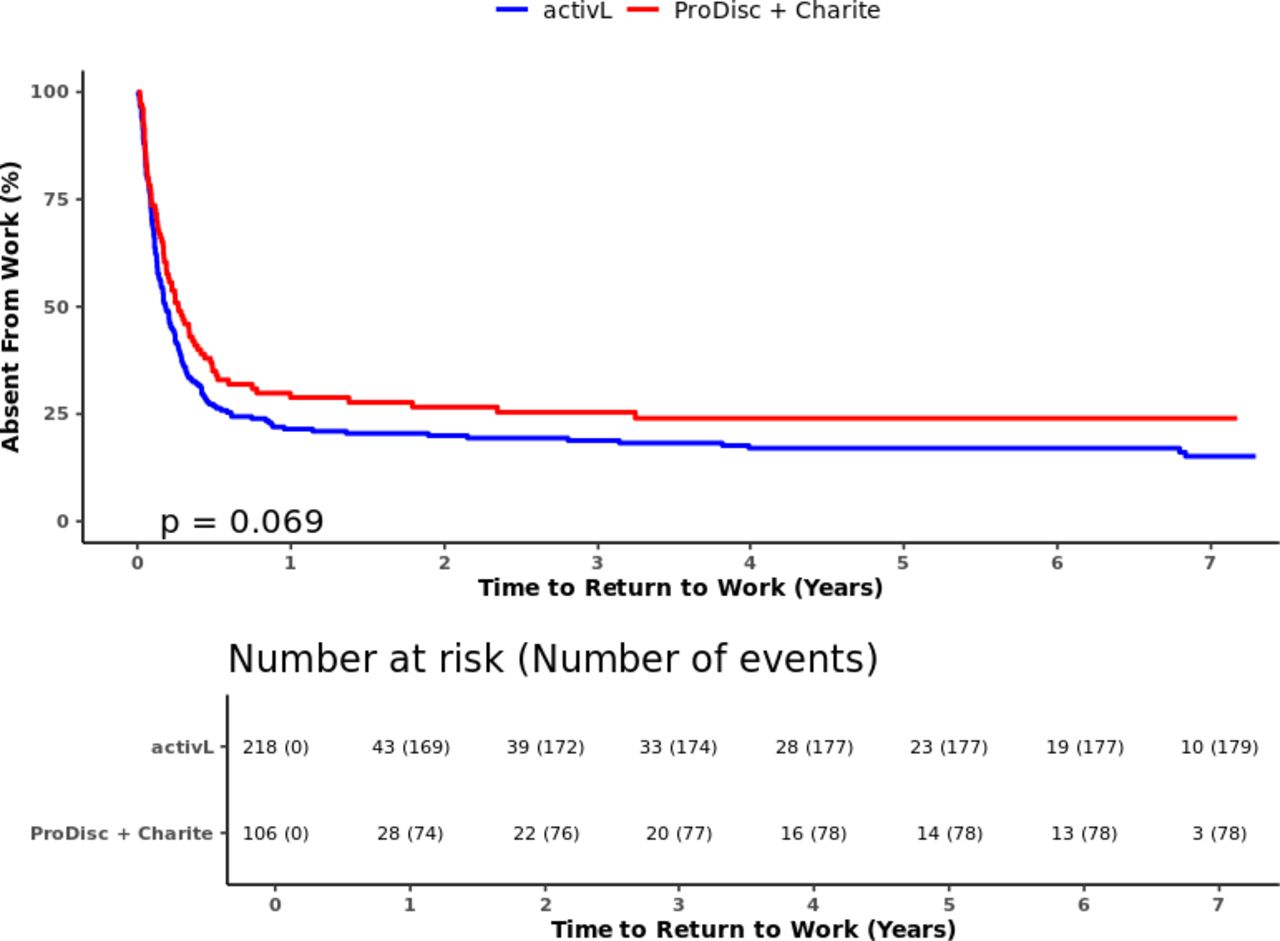
Time to return to work through 7 years for activL versus control (ProDisc-L + Charité). Kaplan-Meier estimate is 82% with activL and 74% with control. Log-rank P value = .069.

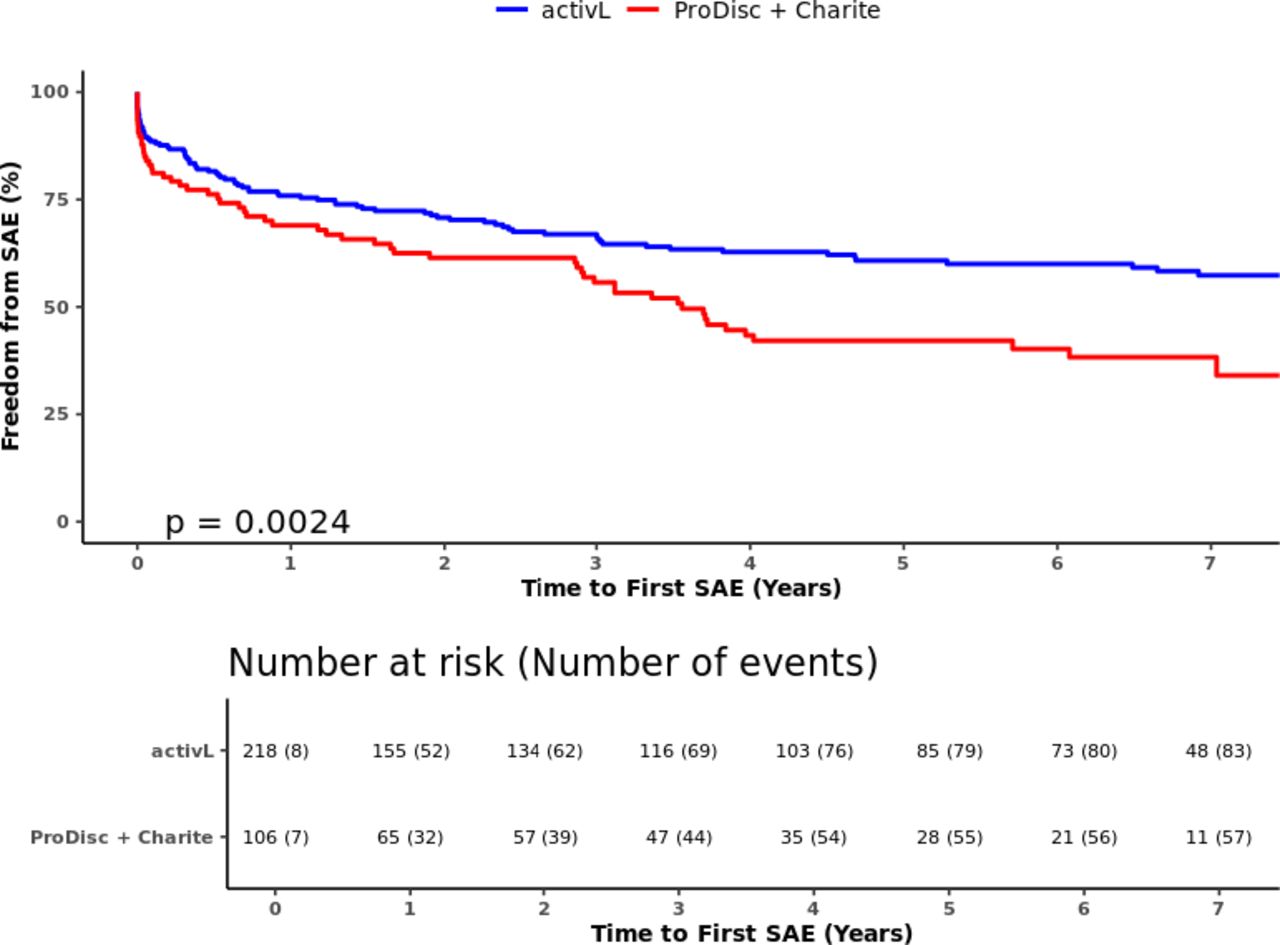
Freedom from serious adverse events through 7 years for activL versus control (ProDisc-L + Charité). Kaplan-Meier estimate is 61.5% with activL and 44.3% with control. Log-rank P value = .0024.
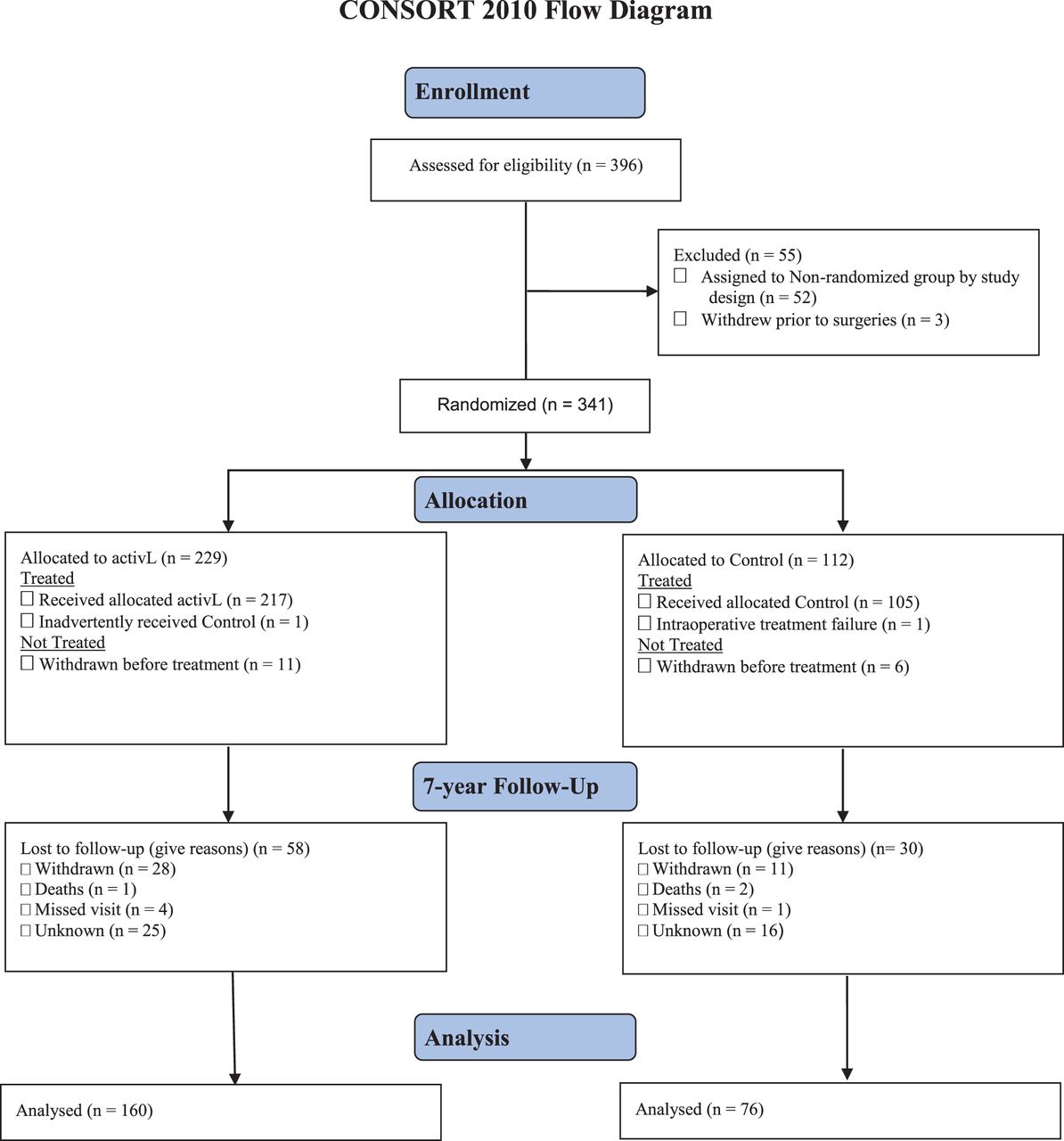
APPENDIX 3. CONSORT FOR ACTIVL VERSUS CONTROL GROUP

CONSORT 2010 flow diagram.
Footnotes
Disclosures and COI: The devices that are the subject of this article were evaluated as part of a US Food and Drug Administration–approved investigational protocol (investigational device exemption) or corresponding national protocol for the treatment of single-level degenerative disc disease of the lumbar spine (L4 to S1) in patients who have been unresponsive to at least 6 months of prior conservative care. Aesculap Implant Systems, LLC (Center Valley, PA, USA), grant funds were received in support of this work. Aesculap Implant Systems, LLC, was involved in the study design and conduct, management of data, and manuscript approval. Relevant financial activities outside the submitted work include board membership, consultancy, expert testimony, payment for lectures, patents, royalties, stocks, and payment for development of educational presentations, travel, accommodations, or meeting expenses.
- This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery. Copyright © 2021 ISASS
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- ABSTRACT
- INTRODUCTION
- METHODS
- RESULTS
- DISCUSSION
- CONCLUSION
- ACKNOWLEDGMENTS
- APPENDIX 1. ACTIVL INVESTIGATIONAL DEVICE EXEMPTION RANDOMIZED TRIAL INCLUSION AND EXCLUSION CRITERIA
- APPENDIX 2. RESULTS OF ACTIVL VERSUS CONTROL (PRODISC-L + CHARITÉ)
- APPENDIX 3. CONSORT FOR ACTIVL VERSUS CONTROL GROUP
- Footnotes
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.