


Abstract
Background Interbody fusion is a widely utilized and accepted procedure to treat advanced debilitating lumbar degenerative disc disease (DDD). Increasingly, surgeons are seeking interbody devices that are large for stability and grafting purposes but can be inserted with less invasive techniques. To achieve these contrary objectives a novel, conformable mesh interbody fusion device was designed to be placed in the disc space through a small portal and filled with bone graft in situ to a large size. This design can reduce the risk of trauma to surrounding structures while creating a large graft footprint that intimately contours to the patient’s own anatomy. The purpose of this Investigational Device Exempt (IDE) trial was to evaluate the perioperative and long-term results of this novel conformable mesh interbody fusion device.
Methods This investigation is a prospective, multicenter, single-arm, Food and Drug Administration and Institutional Review Board-approved IDE, performance goal trial. A total of 102 adults presenting with DDD at a single level between L2 and S1 and unresponsive to 6 months conservative care had instrumented lumbar interbody fusion. Validated assessment tools include 100 mm visual analog scale for pain, Oswestry Disability Index (ODI) for function, single question survey for patient satisfaction, and computed tomography (CT) scan for fusion. Patients were enrolled across 10 geographically distributed sites. Pain/ODI surveys, physical evaluations, and imaging were performed serially through 24 months. Specifically, CT was performed at 12 and, if not fused, 24 months. Independent radiologists assessed CTs for fusion. An independent committee adjudicated adverse events. Patients with complete data at 24 months were included in the analysis.
Results Ninety-six (96, 94% follow-up rate) patients (57.0 ± 12.0 years, 50.0% female, Body Mass Index 30.6 ± 4.9) reported average decreased low back pain from baseline of 45.0 ± 26.6 at 6 weeks and 51.4 ± 26.2 at 24 months. Right/left leg pain reduced by 28.9 ± 36.7/37.8±32.4 at 6 weeks and 30.5±33.0/40.3 34.6 at 24 months. Mean ODI improved 17.1 ± 18.7 from baseline to 6 weeks and 32.0 ± 18.5 by 24 months. At 24 months, 91.7% of patients rated their procedure as excellent/good. Fusion rates were 97.9% (94/96) at 12 months, and 99% (95/96) at 24 months. Mean operative time, estimated blood loss, and length of stay were 2.6 ± 0.9 hours, 137 ± 217 mL, and 2.3 ± 1.2 days, respectively. No device-related serious adverse events have occurred.
Conclusions Clinically significant outcomes for pain, function, fusion, and device safety were demonstrated in this population. Substantial clinical improvements occur by 6 weeks postoperative and continue to improve to 24 months. The successful outcomes observed in this trial support use of this novel device in an instrumented lumbar interbody fusion.
Level of Evidence 3.
Clinical Relevance This reports substantiates that the preliminary 1-year findings published earlier for this investigation are confirmed and the fusion rates and that patient improvements reported are sustained through 2 years.
- minimally invasive spine surgery
- degenerative spondylolisthesis
- lumbar interbody fusion
- transforaminal lumbar interbody fusion
- FDA study
- investigational device exemption
Introduction
Interbody fusion for the treatment of advanced debilitating degenerative disc disease (DDD) of the lumbar spine is widely accepted as reasonable and effective. Interbody fusion, the placement of a biomechanical device or bone graft in the intervertebral disc space, allows for restoration of disc space height, sagittal plane alignment, and weight bearing through the anterior column.1–7 The disc space is a preferred location for fusion as it provides a large surface area for fusion and compressive loads applied to the graft stimulate osteogenesis and fusion. While there are various approaches to achieve lumbar interbody fusions including anterior, lateral, oblique, transforaminal, and posterior, transforaminal lumbar interbody fusion (TLIF) is the most prominent approach worldwide.
Originally introduced to the literature in 1982,8 TLIF was developed to address concerns of the posterior lumbar interbody fusion (PLIF) approach, specifically the degree of neural retraction and potential for nerve root injury, dural tears, and epidural fibrosis.9 More recently, there has been an increased concentration on methods to further minimize potential iatrogenic perioperative morbidity. Minimally invasive surgery (MIS) techniques have led to less muscle damage, bone drilling, and nerve retraction while preserving the advantages of PLIF. However, the MIS-TLIF procedure is still impacted by implant design as the facet joint must often be removed to facilitate interbody cage insertion.10,11
As MIS techniques and systems have developed in other areas of spine surgery,12–15 there has been much focus on how they can be applied to MIS-TLIF. The ideal system would (1) use an access that does not require significant physiologic manipulation or destruction to deliver an implant; (2) provide an implant that can expand in situ to maximize the bone graft footprint to maintain structural stability; (3) have multiple planes of entry to allow for direct contact between the graft and all areas of the endplate interface. We have recently completed an evaluation of such a system (Spineology Interbody Fusion System, Spineology, St. Paul, MN). It is the purpose of this paper to report the 24-month outcomes of this prospective, multicenter, Food and Drug Administration (FDA)-approved investigational device exemption (IDE) study.
Methods
Device
OptiMesh (Spineology, St. Paul, MN) is a biocompatible, radiolucent, porous polyester mesh pouch knitted from polyethylene terephthalate thread (Figure 1). Polyethylene terephthalate material is widely used to make surgical sutures and meshes. In this investigation, the device was placed in the disc space through a small, 8.4 mm portal and filled with bone graft in situ reducing the risk of trauma and dissection requirements of surrounding structures. The portal has since been reduced to 7 mm. The mesh device is pliable, conformable, and strong under tensile and burst forces, as demonstrated through in vitro studies including axial compressive mechanical testing of the filled device’s load-bearing capabilities and overall segmental strength.16 Beyond the mechanical testing, the device performed well in an animal test model to further assess its biomechanical, radiographical and histological properties.17

OptiMesh device filled and unfilled.
Study Design
A prospective, multicenter, single-arm, FDA and Institutional Review Board-approved Investigational Device Exemption (G140140), performance goal trial was undertaken to evaluate the safety and efficacy of a graft containment and reinforcement device in a TLIF application. Patients were enrolled March 2015 through January 2018 across 10 geographically distributed sites in the United States. To be included in the study, patients were required to:
Be in need of intervertebral body fusion to treat DDD at one level between L2 and S1
Be skeletally mature between the ages of 21 and 80 years
Have no greater than grade I spondylolisthesis at the operative level
Have had at least 6 months of conservative treatment
Have a preoperative low back pain (LBP) score ≥40 mm on a 100 mm visual analog scale (VAS)
Have a preoperative Oswestry Disability Index (ODI) score of ≥40
Not have had a previous fusion or total disc replacement at the operative level
Not have 2 or more levels of symptomatic DDD
Not be a current tobacco user (within 30 days of surgery)
Have a body mass index of <40
Not have osteoporosis
See Table 1 for a complete list of inclusion and exclusion criteria.
Study entrance eligibility criteria.
After patients were screened preoperatively (baseline) for all eligibility criteria, they were assessed from surgery through discharge from the hospital and at 6 weeks and 3, 6, 12, and 24 months postsurgery. At each time point, patients received a neurological exam including sensory, motor, and reflexes assessments. Additionally, patients completed the following surveys: low back and lower extremities VAS pain, ODI, 36-item Short Form Health Survey (SF-36), postoperative satisfaction surveys, and work status. Adverse events were assessed continuously throughout the study and were adjudicated for severity and causality by an independent committee. Concomitant medications to manage pain were also captured at baseline and throughout the study.
Outcome Assessments
The primary outcome measure for the study was overall composite success rate of patients at 24 months. The composite score consisted of pain, function, fusion, and patient safety. For success, a patient must have met each of the following requirements:
Pain—Improvement in LBP score evidenced by a 20 mm reduction on a 100 mm VAS when compared to baseline.18–20
Function—Improvement in low back function evidenced by a 15-point decrease of the ODI score compared to baseline.18–20
Fusion—Bridging bone demonstrated on CT imaging.
Safety—Freedom from investigational device-related serious adverse events (SAEs) at the index level through the 24-month study interval. SAEs were defined as an adverse event that requires hospitalization, prolongs or extends the hospitalization, is life-threatening, results in a congenital anomaly/birth defect, or results in death.
Therefore, the study primary endpoint (success) was the proportion of patients who met each of the components of this composite endpoint at 24 months.
Secondary outcomes assessments included individual 24-month analysis of low back VAS pain, lower extremity VAS pain, ODI, fusion at 12 and 24 months, investigational device-related SAEs, study-related AEs, neurological status, index and adjacent level radiographic data, patient satisfaction rated on a 4-item modified Odom Criteria Scale, work status, pain medication use, operative time, estimated blood loss, hospitalization duration, and SF-36.
Radiology
Preoperative x-ray imaging, magnetic resonance imaging, and/or CT were used to confirm eligibility. AP and lateral x-ray images were taken preoperatively, 6 weeks, 3, 6, 12, and 24 months postsurgery. Flexion-extension x-ray images were taken at 6, 12, and 24 months post-urgery per FDA requirement. CT scans were performed at 12 months postsurgery for all patients and at 24 months postsurgery for those not determined to be fused at the 12-month interval. Fusion was defined as bridging bone across the intervertebral space extending from endplate to endplate as assessed on CT scan. All fusion assessments of bridging bone were performed by 2 independent board-certified radiologists. In the event of disparate evaluations, a third radiologist from the core lab adjudicated the case.
The independent radiologists also reviewed imaging for the presence of any of the following: device expulsion, migration, subsidence, radiolucency at the implant/endplate interface, and adjacent level degeneration. For each parameter, the following definitions were applied:
Expulsion: Device having moved outside the disc space
Migration: Greater than 5 mm migration of the implant from its original position
Subsidence: More than 5 mm loss of disc height
Significant radiolucency: Greater than 50% of the implant/endplate interface showing true lucency (ie, black not gray)
Adjacent level degeneration: Greater than 5 mm loss of disc height and > 3 mm translation on flexion-extension views.
All quantitative measurements were performed by an independent, commercial radiographic laboratory. Flexion-extension x-ray images were measured for angulation and translation. Less than 5° of relative angulation and less than 3 mm of translation between flexion and extension x-ray image measurements were predefined as a success. Disc space height was measured on neutral lateral x-ray images prior to discharge and was used as the baseline for subsequent interval measurement comparisons.
Surgical Procedure
Under general anesthesia, either standard open/MIS-TLIF surgical techniques or percutaneous transforaminal approaches to the spine were employed. Surgeons then, using specialized instruments through a small working cannula docked in the annulus, removed a large volume of disc and prepared the endplate. Before proceeding, a contrast-filled compliant balloon (Verify, Spineology, St. Paul, MN) was used to confirm the completeness of the discectomy and proper endplate preparation. Using the working cannula, a mesh device was then introduced into the prepared fusion cavity in its undeployed state and filled with a mixture of allogenic corticocancellous chips and DBM (G2 Dry Mix, Musculoskeletal Transplant Foundation, Edison, NJ), autograft and marrow in situ obtained either locally or from the iliac crest. Iliac crest graft harvest occurred in 77 patients. Using a rotational deployment technique, the device was hypercompacted with the graft mixture to expand and conform the mesh and graft to the cavity, distract the disc space, and stabilize the motion segment by providing a load-bearing construct. The mesh was then detached from the inserter and secured using an integrated cinch string. Finally, posterior supplemental fixation, pedicle screws, were implanted using standard techniques. Surgeons were permitted to use pedicular fixation from the Fortress System (Spineology, St. Paul, MN) or Expedium or Viper Systems (DePuy Synthes Raynham, MA).
Postoperative Care
Postoperative care was prescribed by the treating physician and tailored to the needs of each patient. However, physicians were encouraged to standardized care across investigative sites with the following general instructions to patients:
Avoid driving for a minimum of 1 week following discharge.
Avoid driving while on pain medication.
Ambulate as soon as it is comfortable.
Observe the following lifting restrictions:
Less than 10 pounds for 2 to 4 weeks after the operation.
Less than 20 pounds through 12 weeks after the operation.
Limit bending and twisting for a minimum of 4 weeks after the operation.
Use of a lumbar support brace was optional.
Statistical Analysis
Data collected in this study were documented using summary tables and patient data listings. Continuous variables were summarized using descriptive statistics, specifically the mean and standard deviation are reported. Categorical variables were summarized using frequencies and percentages and 95% exact (Clopper-Pearson) confidence intervals for the true proportions. All analyses of effectiveness and safety were based upon the treated population defined to be all treated patients. All statistical tests were performed at the 0.05 significance level, unless otherwise indicated (SAS v9.2, SAS Institute Inc., Cary, NC, USA).
Results
Baseline and Surgery
A total of 102 patients underwent surgery, and 96 completed the 24-month visit for a 94% follow-up rate. In total, 3 patients exited the study early: 2 relocated out of area and 1 died of pulmonary thromboembolism. Additionally, 3 patients missed a 24-month exam. Of those, 2 were last evaluated at 12 months after the procedure and 1 was evaluated at 12 months and 36 months after the operation; 1 was a study success at 12 months and the remaining 2 were classed as study failures.
At time of informed consent, patients were aged 57.6 ± 12.0 years, were evenly distributed between genders (50.0% female, 50% male), and had a body mass index of 30.6 ± 4.9. Mean ODI score was 53.6 11.7. Average LBP, right leg pain, and left leg pain were 74.1 ± 16.1, 42.9 ± 34.1, and 52.9 ± 32.4, respectively. At baseline, 75.5% (77/102) of patients had 50% back pain and 50% leg pain (defined as VAS scores being within 20 mm of each other). Of the remaining patients, 17.6% (18/102) had predominantly back pain, 4.9% (5/102) had predominantly leg pain, and 2.0% (2/102) had back pain only. In total, 59.8% (61/102) of patients demonstrated listhesis (spondylolisthesis or retrolisthesis) radiographically and 62.4% (63/101) experienced one or more neurological deficiencies at baseline.
Eleven surgeries were performed open, 34 were performed MIS/TLIF, and 57 were performed percutaneously. Forty-one patients had a direct decompression performed during the index procedure and 61 had no direct decompression performed during the index procedure (indirect decompression from the device biomechanical properties). Mean operative time was 2.6 ± 0.9 hours, estimated blood loss was 137 ± 217 mL, and length of stay (LOS) was 2.3 ± 1.2 days. L4-L5 was the most frequent operative level (66.7%) followed by L5-S1 (28.4%), L3-4 (3.9%), and L2-3 (1.0%). Baseline comorbidities are provided in Table 2.
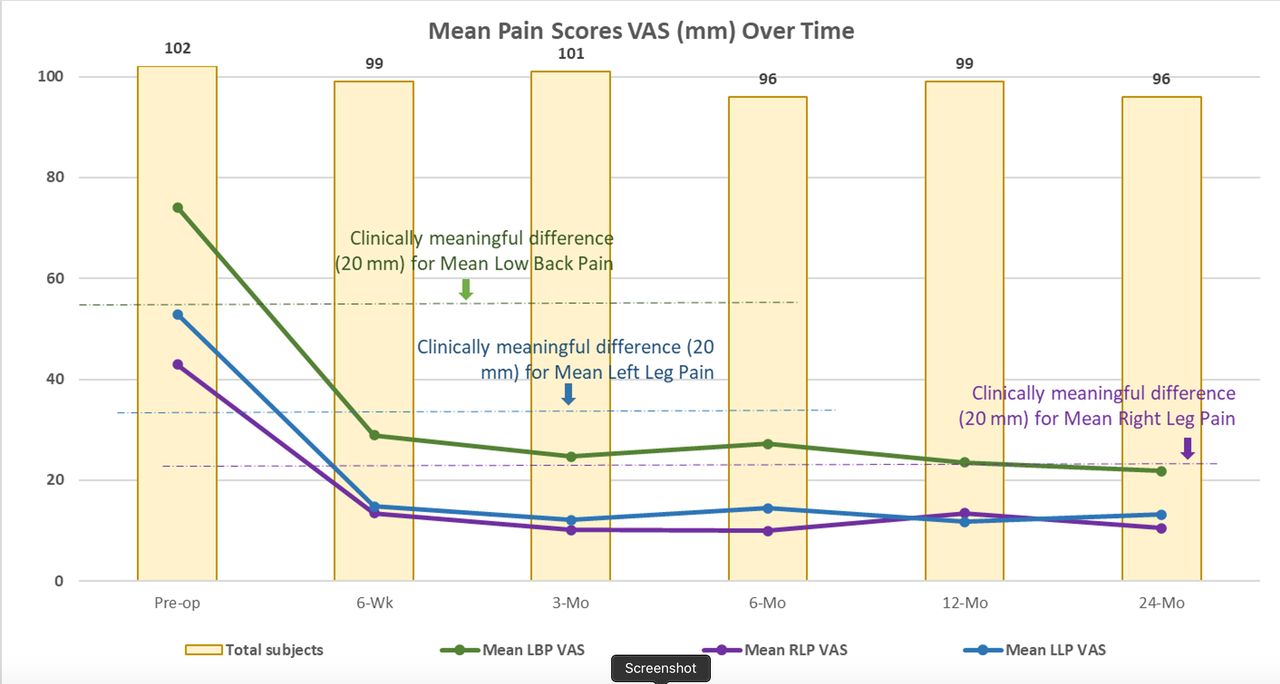
Comorbidities at baseline (N = 102).
Composite Endpoint
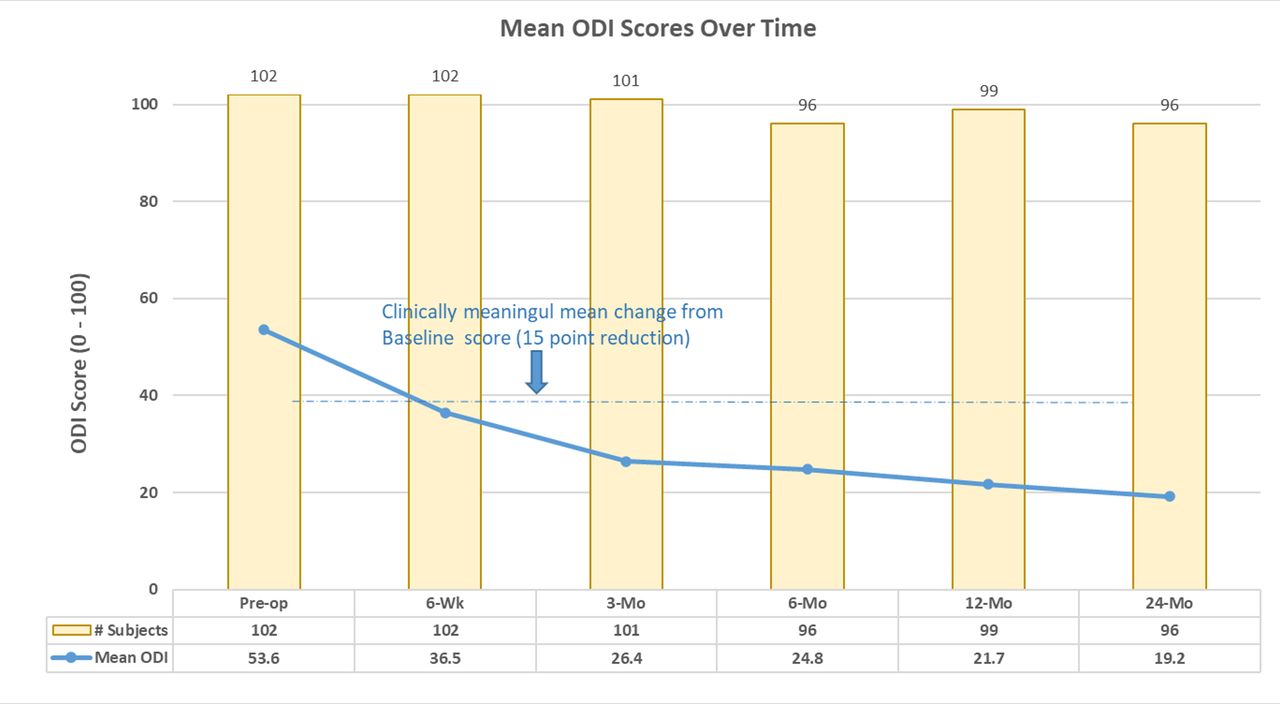
The overall composite success rate of patients at 24 months was 74% (71/96). Individual component success rates were 85.4% (82/96) for LBP, 81.3% (78/96) for function (ODI), 99.0% (95/96) for fusion, and 100% for safety (eg, no investigational device-related SAEs). Average LBP was decreased from baseline by 45.0 ± 26.6 mm at 6 weeks and 51.4 ± 26.2 mm at 24 months. Mean ODI improved 17.1 ± 18.7 from baseline to 6 weeks and 32.0 ± 18.5 by 24 months. See Figure 2 for changes in ODI scores at each study time point.
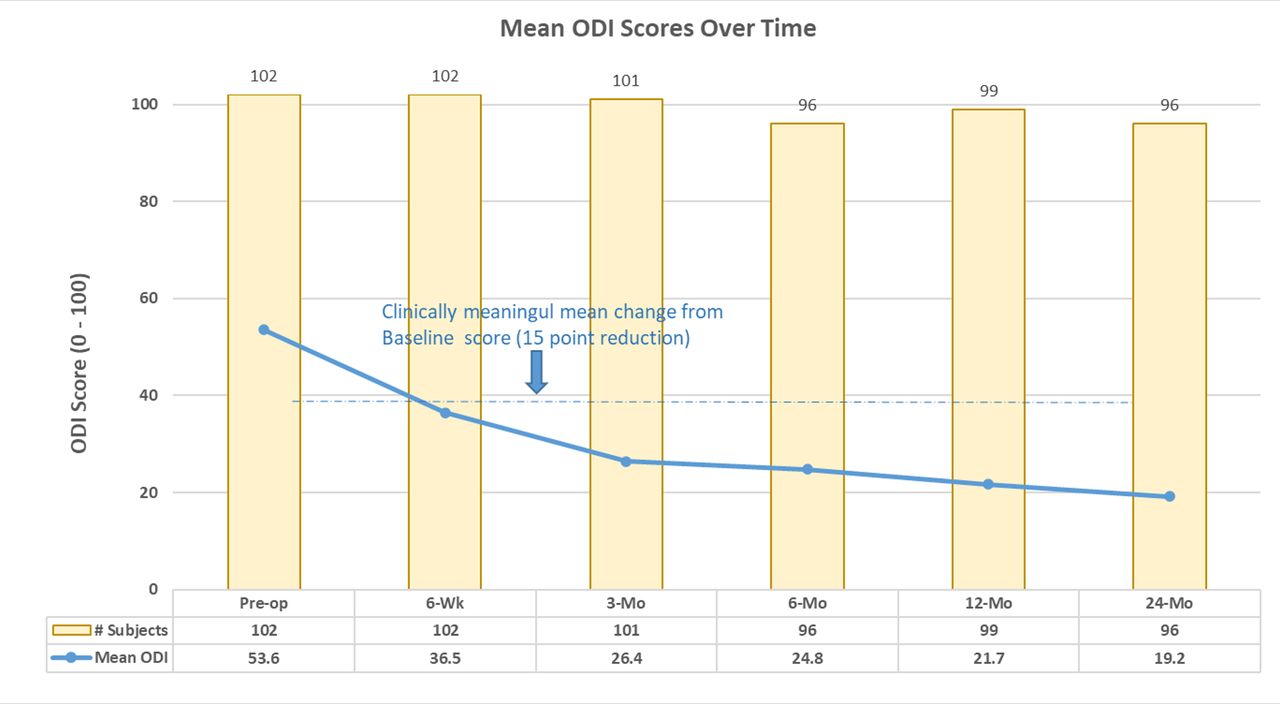
Mean Oswestry Disability Index (ODI) scores through 24 months.
Radiology
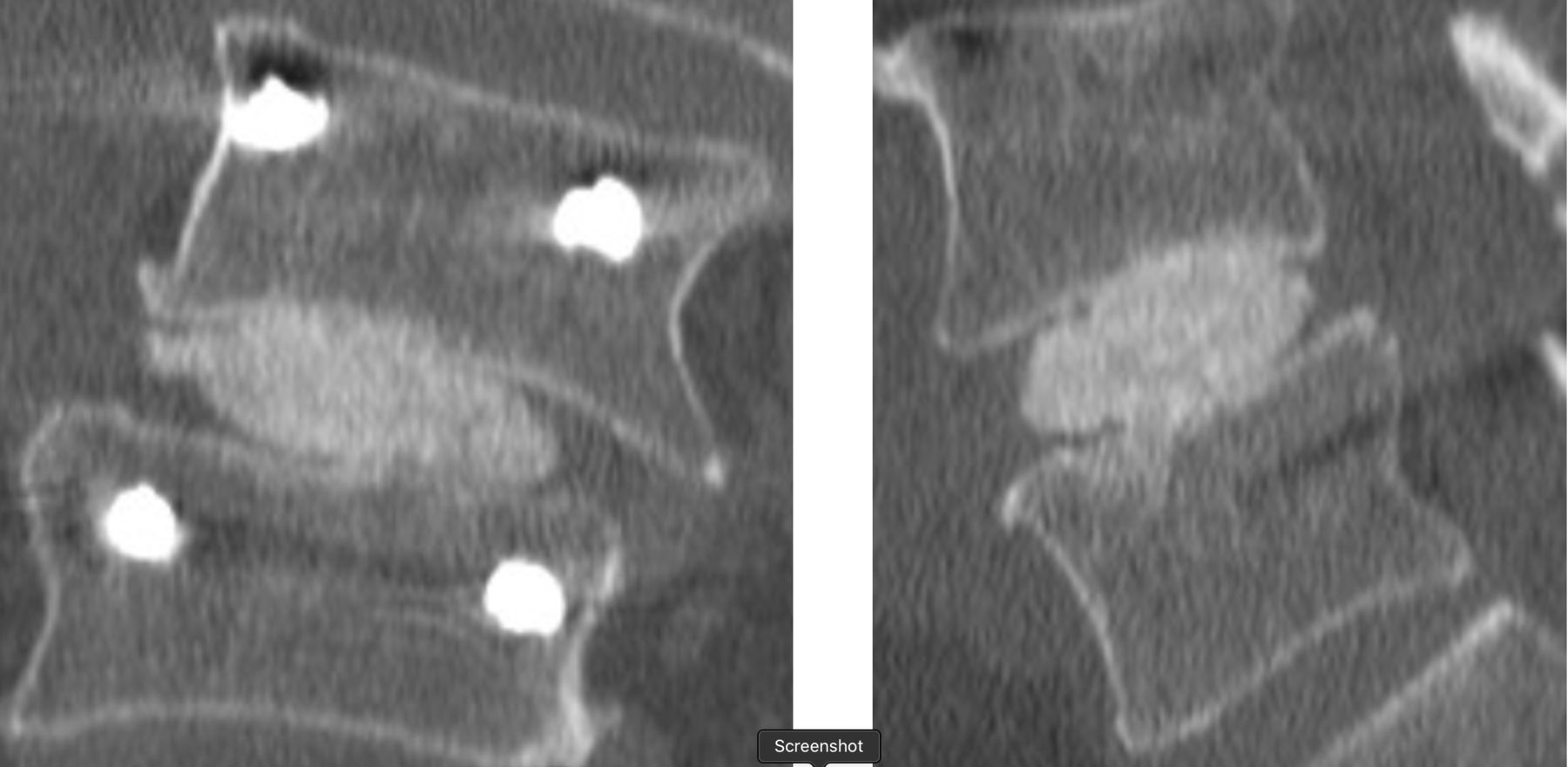
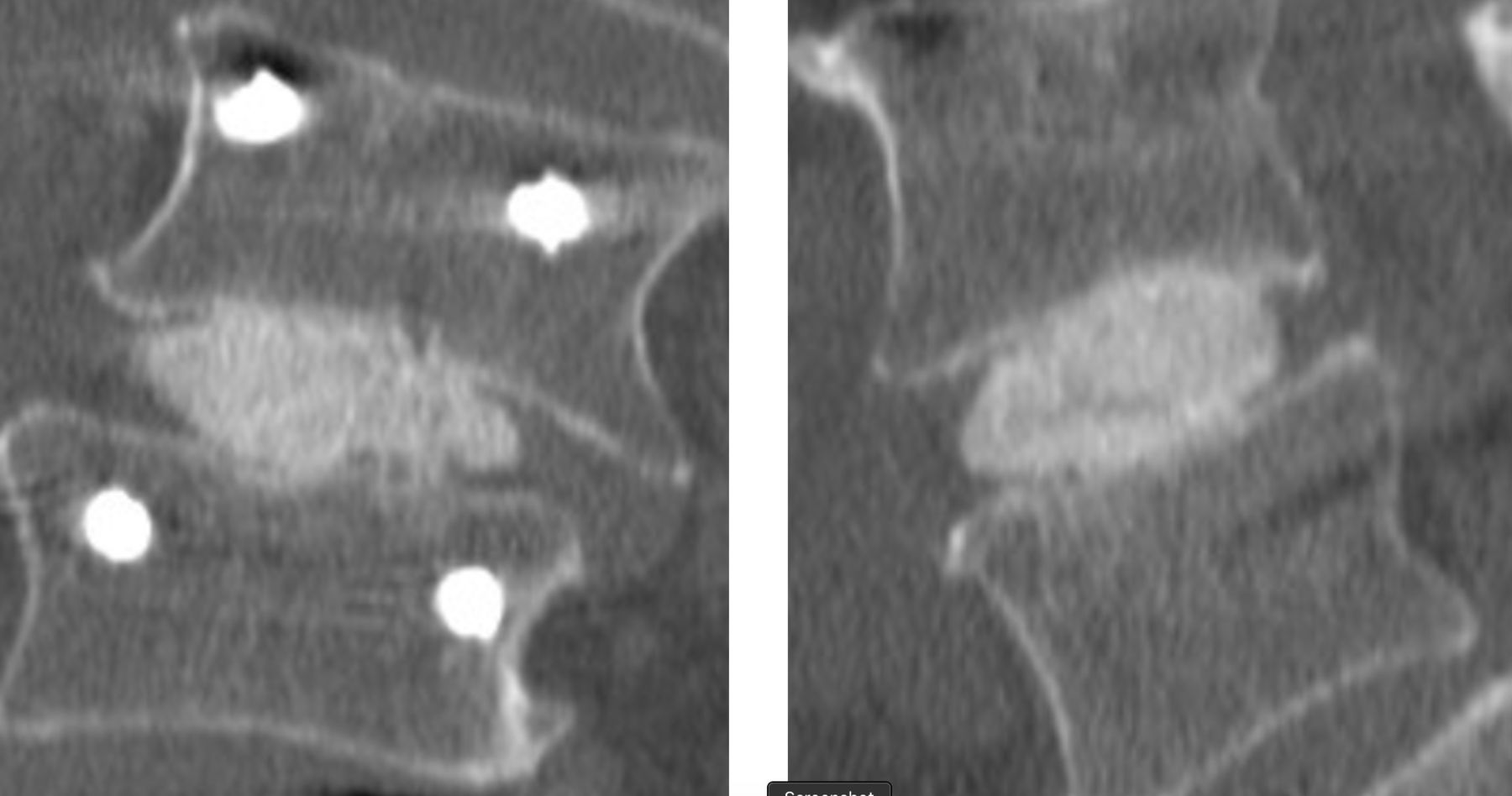
Fusion was observed in 95/96 (99%) of cases at 24 months (Figures 3 and 4). Of the 96 patients evaluated for fusion, a third radiologist at the core lab was called on to adjudicate 6 cases. In all but one case, the adjudicator deemed the segment fused. No (0) device expulsions or retropulsion of disc material during the filling of the device were observed. Measurement of the operative level disc height over time revealed the following: preoperative 6.5 mm ± 2.3, postoperative baseline 9.6 mm ± 2.4, 6 months 7.8 mm + 2.1, 12 months 7.2 mm ± 2.0, and 24 months 7.0 mm ± 1.8. Subsidence was observed in 3 patients with all fused at 12 months. Radiolucency was noted in 2 patients at 12 months. Both fused at 24 months.

Representative computed tomographic images demonstrating fusion in a scoliotic 73-year-old man 12 months after surgery.
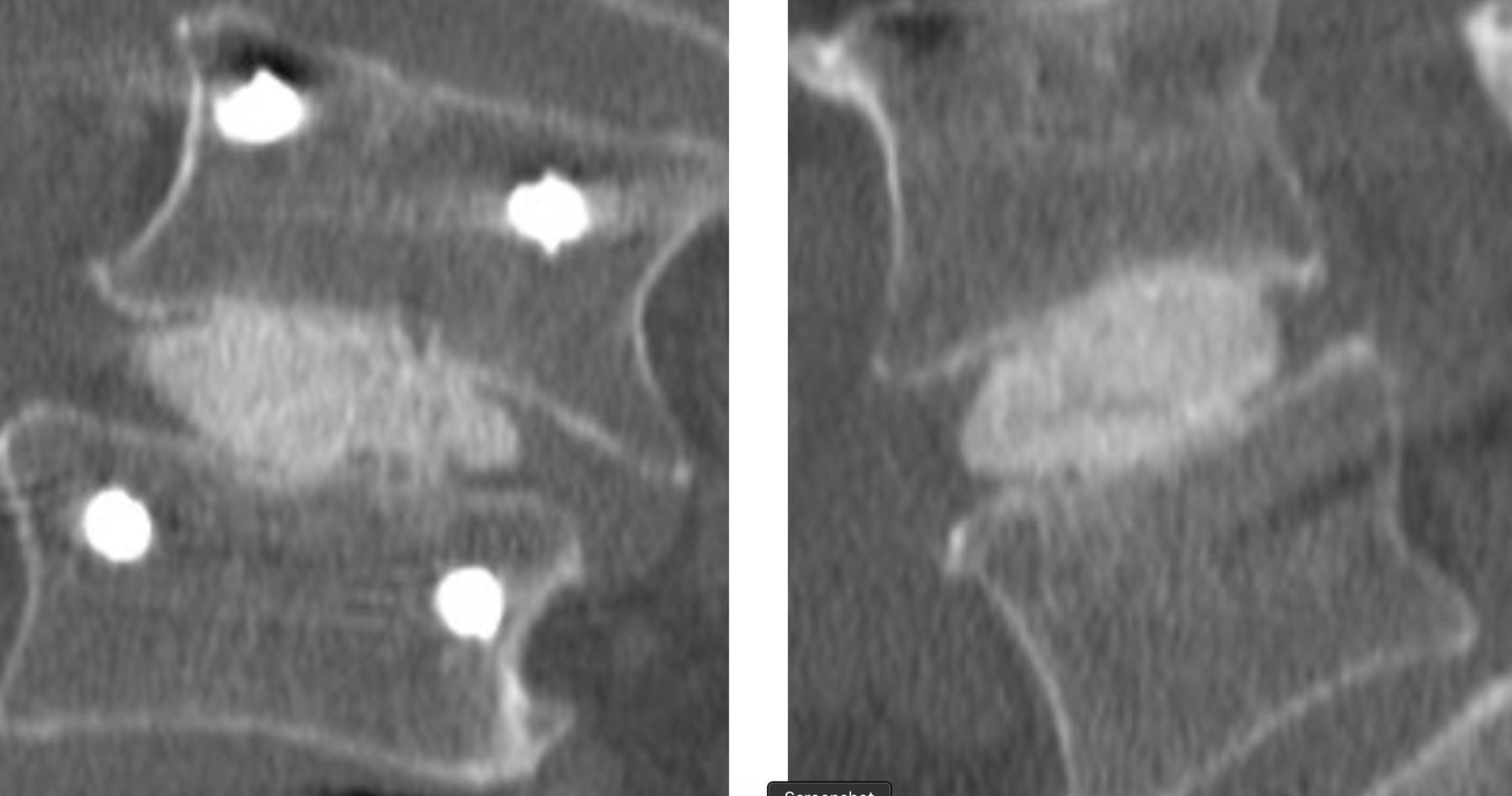
Representative computed tomographic images demonstrating fusion in a scoliotic 74-year-old man 24 months after surgery.
Radiographic adjacent level degeneration was not noted in any patient.
Neurological Changes
There were two reports (2/102) of clinically significant neurologic deficits reported postsurgery. For the first patient, a foot drop was observed on postoperative day 2. Bracing was prescribed but not used by the patient and it was completely resolved by the 6-week exam. For the second patient, after complaining of thigh numbness and foot drop, additional microdecompression at the index level was performed 3 months postoperative and the patient’s symptoms completely resolved.
VAS Pain
As previously reported, LBP scores decreased from baseline by 45.0 ± 26.6 mm at 6 weeks and 51.4 ± 26.2 mm at 24 months. Right/left leg pain reduced by 28.9 ± 36.7/37.8 ± 32.4 at 6 weeks and 30.5 ± 33.0/40.3 ± 34.6 at 24 months. See Figure 5 for VAS LBP and leg pain scores across the study.
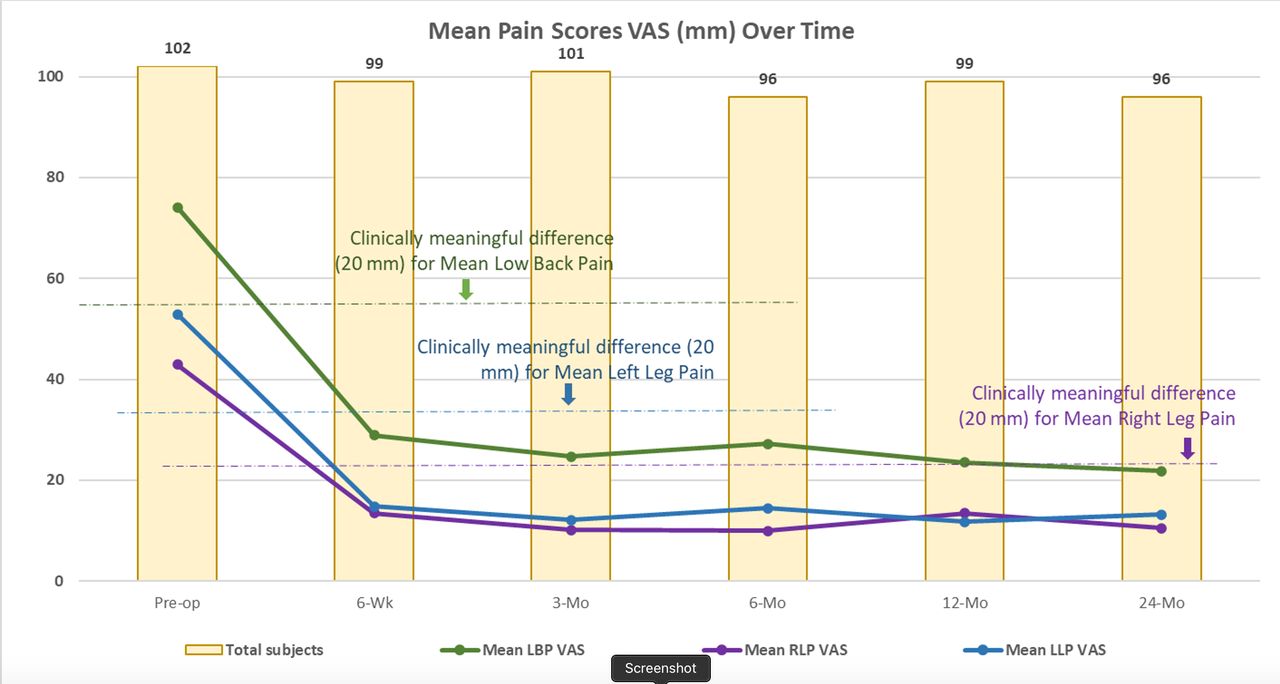
Mean low back and leg pain visual analog scale (VAS) scores over time through 24 months.
Patient Satisfaction
Patients were asked to rate their satisfaction with the surgical procedure as excellent, good, fair, or poor. At 24 months, satisfaction was high with 91.7% rating their procedure as excellent or good (64.6% excellent, 27.1% good, 6.3% fair, and 2.1% poor).
Work Status
At baseline, 52.9% (54/102) patients were working. Of the 47.1% (48/102) not working, 16.7% (17/102) were due to back pain/injury, 23.5% (24/102) were retired, 1.0% (1/102) were students, and 5.9% (6/102) were not working for other reasons. At 6 weeks postoperative, 30.3% (30/99) of patients were working. This rate was increased at 3 months from surgery (40.0%, 40/100) and was further improved at 6 months postoperative where 51.5% (50/97) of patients were working. One year from their index procedure, 50.5% (50/99) of patients were working and a similar rate of employment was observed at 24 months postoperative (47.9%, 46/96). Of importance, the rate of patients not working due to back injury/pain decreased from 16.7% (17/102) at baseline to 8.3% (8/96) at 24 months, representing a 50% improvement for that group.
Pain Medication
At baseline, 80.4% (82/102) of patients were using medication to manage their back pain. Of those, 49.0% (50/102) were using narcotic medication and 69.6% (71/102) were using non-narcotic medication to manage their LBP. Narcotic use to manage LBP at 24 months was decreased to 17.7% (17/96). Non-narcotic pain management medication use for LBP decreased to 59.4%. It is important to note medication use reporting includes both daily and PRN use. This method of reporting is consistent from the preoperative visit through 24 months.
SF-36
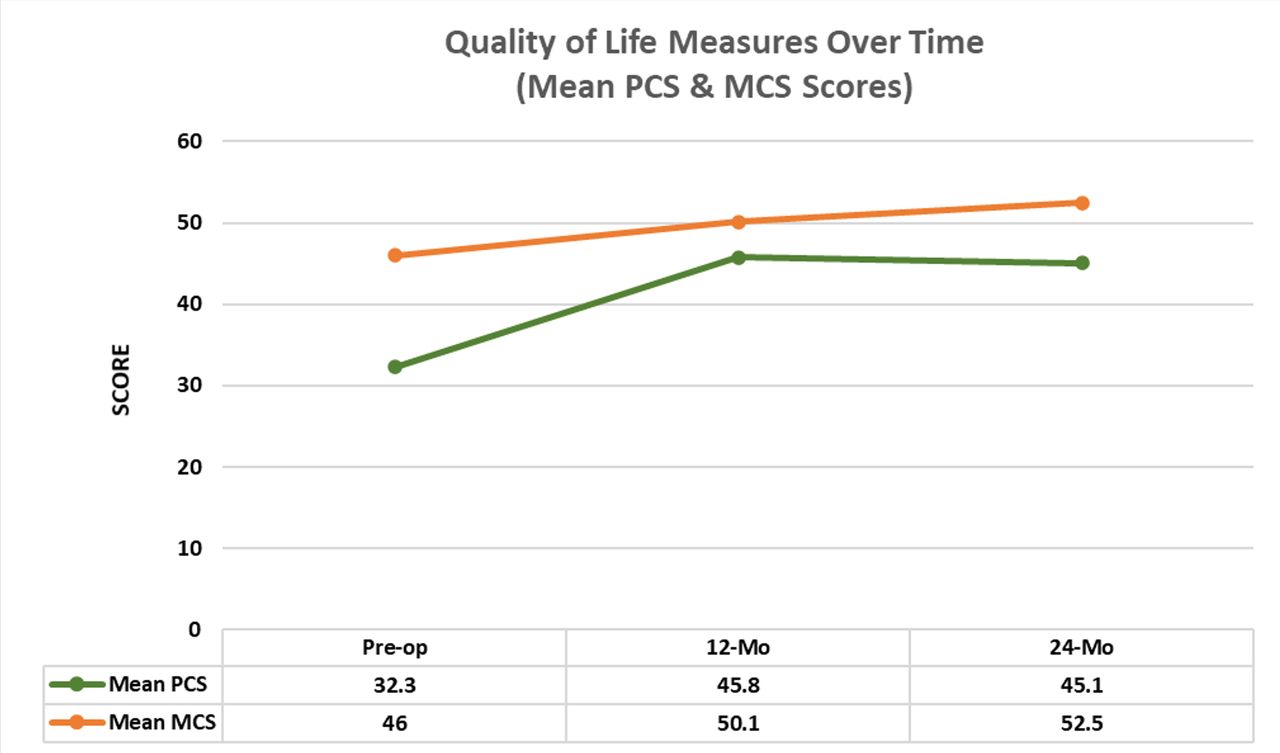
At baseline, the PCS summary score of the study population was 32.3 ±6.4, below the generalized US population mean of 50, as expected. PCS scores increased to 45.8 ± 11.0 at 12 months and 45.1 ± 10.5 at the 24-month timepoint. The study population MCS was also below the norm at baseline with a mean MCS score of 46.0 ± 11.0. Improvement at 12 and 24 months postoperative is observed in the mean 12-month MCS score of 50.1 ± 11.8 and 24-month MCS of 52.5 ± 10.6. See Figure 6 for SF-36 scores over time.

SF-36 physical and mental component scores over time.
Serious Adverse Events
The independent Clinical Events Committee adjudicated 15 adverse events to be study-related SAEs. Of the 15, 6 required secondary surgical intervention at the index level, 3 required surgical intervention at an adjacent level, 2 hematomas required evaluation, 1 dural leak required intraoperative repair, and 3 required hospitalization without surgery. One of the 3 patients requiring hospitalization without surgery expired resultant of a pulmonary thromboembolism. Two of the 6 patients requiring secondary surgical intervention at the index level had the device explanted, one resultant of improper device placement during the index procedure and one for ongoing back/leg pain. Table 3 presents a tabular listing of these events. None of the SAEs were determined to be device related.
Study-related serious adverse events.
Device-Related Adverse Event
There was a single device-related event that was not deemed to be an SAE by the Clinical Events Committee and the investigator. This event occurred in a 31-year-old man with a previous adjacent level disc replacement. The patient reported increased low back and right leg pain at 3 months after the operation. X-ray imaging revealed a L4 vertebral body endplate fracture that was treated conservatively.
Discussion
Here we report the 24-month outcomes of a prospective investigational device exempt single-arm study evaluating a novel conformable mesh interbody fusion device for the treatment of DDD with or without spondylolisthesis requiring single-level fusion. The results demonstrate favorable patient-reported outcome measures as well as exceptionally high fusion rates, and an absence of serious device-related complications.
The primary outcome of this study consisted of a composite endpoint, combining patient-reported outcomes, radiographic outcomes, and safety outcomes into a single endpoint. At 24 months after surgery, we found that overall composite success rate was 74%, corresponding to all of the following: a significant improvement in back and/or leg pain, improved quality of life scores, successful radiographic fusion, and absence of SAEs for the majority of patients. The inclusion and reporting of a composite endpoint for overall success is relatively rare for lumbar interbody fusion studies. However, in the small number of lumbar studies that does report a composite endpoint consisting of clinical, radiographic, and patient-reported outcomes, overall success rates range from 18% to 76%,21–25 placing the results of this study at the higher end of the spectrum. These results also compare favorably to other novel spinal devices, such as for cervical arthroplasty and fusion which range from 60% to 80% overall success on a composite endpoint.26–29
Individual outcome measures showed that 92% of patients rated their satisfaction as good or excellent at 24 months postprocedure. The overall fusion rate was 99% at 24 months following review by multiple independent radiologists. These are excellent results and compare favorably to other studies evaluating fusion rates with variable interbody fusion techniques, ranging from 70% to 95%.30 With regard to quality of life, patients reported significant improvements in back pain and leg pain VAS scores as well as ODI scores at the initial 6-week postoperative visit. Previous reports on the conformable mesh interbody device demonstrated good outcomes at 12 months postoperatively,31 but it remained uncertain how durable those outcomes might be. Here we demonstrate that improvements in pain and quality of life scores persisted up to the 24-month period. Consistent with previous reports on minimally invasive interbody fusion,32,33 the results of this study offer encouraging findings relating to clinical outcomes.
In terms of complications, there were no serious device-related complications. Two patients had postoperative neurologic deficit, one of which required microlumbar decompression at the index level. Both patients had resolution of symptoms and return to neurologic baseline within 6 weeks and 3 months, respectively. The total incidence of adverse events, including hematomas, durotomy, medical complications, and index level or adjacent level disease requiring additional surgery was 14.7%. This is comparable to other MIS-TLIF studies, with one large review of 513 patients reporting an overall complication rate of 15.6%.34
Lumbar interbody fusion is a critical tool for the treatment of symptomatic lumbar DDD in the setting of unremitting back pain and/or radiculopathy.35,36 Multiple techniques have been developed and are now well accepted for achieving interbody fusion, with overall similar results in terms of patient outcomes and fusion rates.37–40 Open surgical techniques, however, require a certain degree of tissue manipulation, muscle injury, bony drilling, and retraction of neural tissue, all of which can contribute to increased patient-reported pain, operative time, blood loss, and length of stay.41–43 Technological advances have allowed for the adoption of MIS techniques which minimize tissue disruption and may decrease surgical morbidity.12–14 Endoscopic and percutaneous techniques, which initially were used for simple lumbar decompressive surgery and discectomy, have now been expanded to interbody fusion, achieving results similar to TLIF, but with smaller incisions and less tissue manipulation.44
Conventional open and MIS-TLIF surgery, requires a certain degree of facet removal for safe placement of a rigid interbody cage which must be passed adjacent to the exiting and traversing nerve roots. Resultant of the device being placed through a small portal and the subsequent deployment in situ, the surgeon can reduce the impact on surrounding anatomical structures and tissue. Placing the device through a percutaneous technique to access the interbody space, further allows for preservation of the facet joint, which offers the promise of expediting postoperative recovery and better preserving the integrity of the posterior column.33 The use of a novel conformable mesh, as presented here, allows for minimizing bony removal while still achieving a low risk of injury to the nerves or thecal sac because of the small profile of the device prior to being deployed in the interbody space. Despite the small profile, the device can be “expanded” which allows for robust structural support, an increase in foraminal size, and ultimately fusion across the disc space. The biomechanical properties of the conformable mesh device have been studied as an interbody system and have shown that with posterior fixation, such as pedicle screws, the device is able to withstand physiological loads with minimal deformation.45 These features allow for such an expandable interbody mesh system to be deployed though an MIS approach could provide considerable advantages to other methods.
The limitations of this evaluation are the relatively small number of subjects and that it was a single arm trial which did not compare directly to other established methods for achieving interbody fusion. Despite these factors, demonstration of the positive outcomes including high rates of improved patient self-reported outcomes, excellent fusion rates, and strong safety profile for approximately 100 patients, identifies OptiMesh as a viable option for interbody fusion. However, as with many advances in medicine, adoption of this technique must occur in the context of the abundant literature pertaining to interbody fusion.
Conclusion
This study demonstrates that use of this novel interbody fusion device in the treatment of patients with single-level degenerative disc disease, with or without spondylolisthesis, successfully improved patient symptomology in terms of pain relief and daily activity functions and that they are improved or sustained over time through 2 years. The tissue-sparing surgical technique, high rates of fusion, and a strong safety record, coupled with favorable patient self-reported outcomes, support adoption of this unique technology.
Footnotes
Funding Marcus Stone discloses support for the present article (eg, funding, provision of study materials, medical writing, etc) from Spineology. No other author(s) received financial support for the research, authorship, and/or publication of this article.
Financial Disclosures and Conflicts of Interest John Chi discloses consulting fees from DePuy Spine and Stryker/K2M. Pierce Nunley discloses royalties or licenses from Stryker, Zimmer Biomet, and IMSE; consulting fees from Spineology, Stryker, IMSE, Integrity Implants, Kuros, NEO Spine, and Simplify Medical; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or education events from Instrinsic Therapeutics and Organogenesis; and stock or stock options from Spinelogy and Surgalign. Stephane Lavoie discloses royalties or licenses from Spineology and consulting fees from Choice Spine and Spineology. Yi Lu discloses grants from the Stepping Strong Foundation (paid to institution); consulting fees from DePuy/Synthes; and stock or stock options in AxioMed LLC. Marcus Stone discloses support for the present article (eg, funding, provision of study materials, medical writing, etc) from Spineology; grants or contracts from Wright Medical (paid to institution); consulting fees from Spineology and Organogenesis (paid to Marcus Stone) and from Simplify Medical, Zimmer Biomet, and Spineology (paid to institution). All remaining authors report no relevant disclosures.
- This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery. Copyright © 2021 ISASS. To see more or order reprints or permissions, see http://ijssurgery.com.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.