


Abstract
Background The purpose of this study was to determine the indications for implantation of the coflex device (Paradigm Spine, LLC, New York, New York), assess long-term complications, and evaluate the long-term clinical outcomes of patients.
Methods A total of 127 patients underwent placement of a coflex implant for various indications by one orthopaedic spine surgeon. The mean follow-up was 6.3 years. The original indications for implantation were determined based upon the data provided in the case report forms. Preoperatively and postoperatively, patients were asked to grade their low-back and leg pain using the visual analog scale (VAS) and the pain severity scale: no pain (0), mild pain (1), moderate pain (2), or severe pain (3). Patients were queried about their satisfaction with the surgical procedure. Follow-up radiographs were taken to determine any device-related issues.
Results The most prevalent diagnoses for implantation were spinal stenosis (19.7%) and spinal stenosis with lumbar disc herniation (35.4%). The mean severity of low-back pain was decreased by 33% (from moderate to mild) at the 2-year follow-up (P < .001) and at the 5-year follow-up (from moderate to mild, P < .001). The mean severity of leg pain was decreased by 66% (from severe to mild) at the 2-year follow-up (P < .001) and at the 5-year follow-up (from severe to mild, P < .001). At the mean follow-up period of 6.3 years, a patient satisfaction query demonstrated that 7% were unsatisfied, 46% were satisfied, and 46% were very satisfied with their clinical outcome. Based on the follow-up radiographs, 92 of patients had no devicerelated issues and 8% had device-related issues.
Conclusion The data provided have demonstrated that the coflex implant provides pain relief for patients with low-back pain and leg pain. The most common indications for implantation were spinal stenosis and spinal stenosis with lumbar disc herniation. There were very few device-related complications.
Clinical Significance Using coflex is a safe and viable option in the selection of instrumentation for spinal stabilization.
- Interspinous “U”
- coflex
- interlaminar-interspinous implant
- spine arthroplasty
- spinal stenosis
- long-term outcomes
INTRODUCTION
Spinal stenosis with neurogenic claudication was first described by Verbiest in 1954.1 Recently, interlaminarinterspinous devices and dynamic stabilization devices have been approached as an alternative to fusion surgery in treating spinal stenosis. These devices are designed to unload the facet joints while increasing spinal canal diameter and restoring foraminal height. In order to be an effective implant for the treatment of spinal stenosis and not subsequently cause disc pathology at adjacent levels, an interlaminar-interspinous or dynamic stabilization implant should decrease intradiscal pressure (IDP) at the instrumented level while leaving the adjacent segment IDP unaffected.2 The first device to tackle this disease process was envisioned as a silicone dumbbell-shaped device to be inserted between the spinous processes.3 This posterior interspinous dynamic stabilization device was biomechanically tested to reduce deflection as well as reduce IDP at the level the implant was placed. Since then, only one device has been FDA approved and 3 are under investigation.4–8 The dynamic stabilization device for neurogenic claudication secondary to spinal stenosis that is currently FDA approved (X-STOP Interspinous Process Decompression System, Medtronic Sofamor Danek, Inc., Memphis, Tennessee) has been proven to be safe and effective.9–14 Additionally, it has been shown to reduce IDP at the instrumented segment as well as leave the adjacent IDP unaffected10 and increase the spinal canal diameter and area.11 However, the overall range of motion (ROM) is significantly reduced using the X-STOP, especially from flexion to neutral.9
The coflex interlaminar-interspinous implant (Paradigm Spine, LLC, New York, New York) is a “U” shaped implant (Figure 1) manufactured by a milling process of titanium alloy, which has been engineered to resist normal physiologic loads in the spine.15 The device is available in heights of 8, 10, 12, 14 and 16 mm with two wings on the upper and lower ends. The device is positioned using a muscle-sparing posterior midline approach and is inserted between adjacent spinous processes of the lumbar spine. Before implantation, the interspinous ligaments and their bony attachment are removed, while mobilizing and protecting the supraspinous ligament for reconstruction.15, 16 Once implanted, the lateral wings are crimped towards the spinous process to improve fixation.

The coflex dynamic stabilization device.
Biomechanical testing of the interlaminar-interspinous implant has demonstrated that it increased ROM in flexion, axial rotation, and lateral bending while causing a decrease in the amount of extension.17 Furthermore, the investigational device caused no change in IDP in flexion, axial rotation, or lateral bending but it did decrease the traditional IDP gain in extension.
Previous clinical trials of patients being treated for spinal stenosis with or without herniated discs have demonstrated that 74% of patients implanted with the interlaminar-interspinous device reported excellent outcomes.15 Other studies have shown that implantation is equally effective in patients with lumbar spinal stenosis with or without herniated discs.18 However, these studies have demonstrated that the interlaminarinterspinous implant is not as effective in reducing pain in patients with iatrogenic dynamic instability secondary to radical disc resection.18 Many authors have suggested that implantation with the interlaminarinterspinous implant is a safe alternative for patients that want a less invasive surgical treatment and do not want to undergo major fusion surgery.19, 20
Little is known concerning the true indications for implantation of the investigational device. Prior to our study, the most reliable clinical outcome study completed is a 1-year follow-up study of patients treated for spinal stenosis, angular instability, or translational instability with the implant. This study demonstrated that there was significant improvement in the clinical outcome in patients that had an interlaminar-interspinous implant placed as compared to traditional posterior lumbar interbody fusion (PLIF).21 Radiographic evaluation of these patients not only demonstrated that the investigational device decreased ROM significantly less than PLIF at the level of implantation, but it also did not increase the ROM at adjacent segments.
The purpose of this study was to determine the indications for implantation of the interlaminarinterspinous implant, the long-term adverse events from implantation, and to assess the long-term clinical outcomes of the study patients implanted with the interlaminar-interspinous device from a single spine surgeon's practice.
METHODS
A total of 127 patients underwent placement of an interlaminar-interspinous implant (coflex, Paradigm Spine, LLC, New York, New York) by one orthopaedic spine surgeon. The patients’ mean age at the time of surgery was 54.8 years (range: 21 to 81 years old). There were 75 females and 52 males. The mean follow-up was 6.3 years (range: 3 months to 12 years). Patients were seen both preoperatively and postoperatively by the same operating orthopaedic spine surgeon. The operating surgeon compiled a case report form (CRF) on every patient enrolled in the study. Retrospectively, the original indications for implantation were determined collectively by one attending orthopaedic spine surgeon and two orthopaedic spine research fellows, using the data provided in the CRFs.
Preoperatively and postoperatively, patients were asked to grade their low-back and leg pain using the pain severity scale: no pain (0), mild (1), moderate (2), or severe (3). Additionally, the visual analog scale (VAS) was employed to assess leg and back pain. Patients were also queried regarding their satisfaction with their operative procedure using the following scale: unsatisfied, satisfied, and very satisfied with respect to their clinical outcome.
Postoperatively, patients received radiographic analysis at their follow-up visit. The operating surgeon assessed the patients’ radiographs for any device-related issues. Adverse events from implantation were determined retrospectively by the operating surgeon.
Statistical Methods
Student's t test was performed comparing the preoperative and postoperative grades for the low-back pain severity scale, the leg pain severity scale, and the VAS in patients at the 2-years follow-up and 5-year follow-up visits. Statistical significance was defined as P < .05.
RESULTS
While analyzing the CRFs of 127 patients, complete data on 99 patients demonstrated that the interlaminarinterspinous device was implanted in patients primarily due to the following diagnoses at the level of implantation: spinal stenosis (19.7%), spinal stenosis with lumbar disc herniation (35.4%), and lumbar disc herniation (7.9%). Figure 2 illustrates the other diagnostic inclusion criteria and their respective overall percentages.
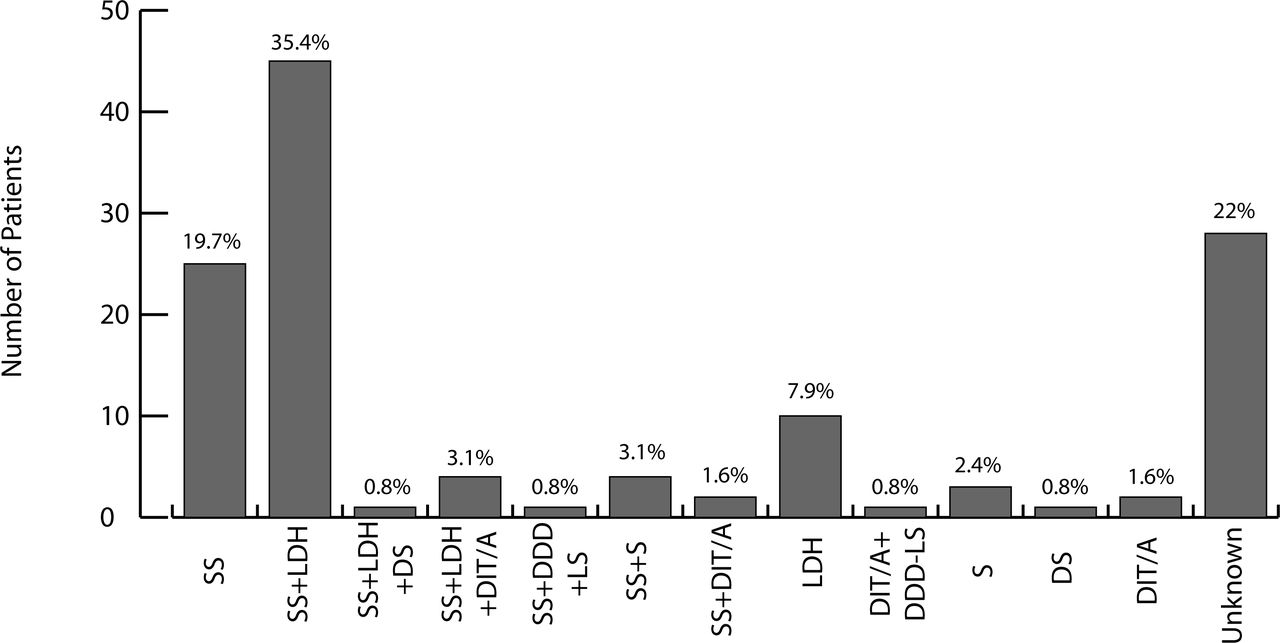
Indications for Implantation: SS - Spinal Stenosis; LDH - Lumbar Disc Herniation; DS - Degenerative Spondylolisthesis, DIT/A - Dynamic Instability- Translatory (≥2 mm) or Angular (≥3 degrees); DDD-LS - Degenerative Disc Disease of Lumbar Spine; S - Scoliosis.
One hundred patients received 1-level insertion and 27 patients received 2-level insertion. A total of 154 implants were inserted with the following breakdown of the level of insertion: 1 implant at T12-L1, 2 implants at L1-L2, 33 implants at L2-L3, 38 implants at L3-L4, 69 implants at L4-L5, and 11 implants at L5-S1. Table 1 lists procedures performed at the level of insertion in addition to implant insertion. The mean operative time was 88 minutes for 1 implant (n = 39) and 98 minutes for 2 implants (n = 5), with a mean estimated blood loss of 285 mL for 1 implant (n = 46) and 333 mL for 2 implants (n = 7).
Additional Surgical Procedures
The mean severity of low-back pain was able to be calculated based on 123 patients, and a graph was constructed (Figure 3) which demonstrates that the severity of low-back pain was decreased by 33% (from moderate to mild) at the 2-year follow-up (P < .001, n = 106) and remained lowered by 33% (from moderate to mild) at the 5-year follow-up (P < .001, n = 77). Patients with the longest follow-up had the higher pain scores while those with the shortest follow-up had the lowest severity of low-back pain, but this finding was found to be insignificant (P > .05). VAS back pain scores also decreased at the 2-year follow-up (from 6.4 to 1.8, P = .002, n = 6) and maintained that reduction at least 5 years after surgery (from 6.4 to 1.8, P = .02, n = 4). The mean severity of leg pain (Figure 4) was calculated and the data demonstrate that leg pain was decreased by 66% (from severe to mild) at the 2-year follow-up (P < .001, n = 17) and remained decreased by 66% (from severe to mild) at least 5 years after surgery (P < .001, n = 77). VAS leg pain scores decreased at the 2-year follow-up (from 6.6 to 0.8, P < .001, n = 6) and at least 5 years post-surgery (from 6.5 to 0, P < .001, n = 4).

Low-back pain severity, preoperative and postoperative: 0 - no pain; 1 - mild pain, 2 - moderate pain; 3 - severe pain.

Leg pain severity, preoperative and postoperative: 0 - no pain; 1 - mild pain, 2 - moderate pain; 3 - severe pain.
Overall patient satisfaction scores were tabulated and it was determined that 7% (n = 9) were unsatisfied, 46% (n = 58) were satisfied, and 46% (n = 58) were very satisfied with their clinical outcome. Concerning the patients that were unsatisfied, 8 patients had postoperative back pain and only 3 of them rated the back pain to be worse. All 9 of the unsatisfied patients had postoperative leg pain and only 2 of them rated the leg pain to be worse.
Ninety-nine (99) patients had follow-up radiographs. The adverse events were categorized into the following groups with their respective percentages (Figure 5): no device-related issues (93%) and device-related issues (7%). The patients with device-related issues were categorized into the following subgroups: broken “U” portion of implant (0%); broken wing of the implant (1%); displaced “U” portion of implant less than 5 mm (2%); displaced “U” portion of implant more than 5 mm (2%); displaced “U” portion of implant with unknown distance (1%); removed implant (2%); spinous process fracture (0%); and bone-implant interface remodeling (0%). Of the 2 patients that had their implant removed, the first patient did not have a good indication for surgery and had a canal that was too narrow and straight; the second patient elected to undergo a 2-level fusion one year after implantation with the interlaminarinterspinous device.

Number of device-related adverse events as determined by radiograph (n = 99).
DISCUSSION
Until recently, surgical treatment for spinal stenosis, degenerative spondylolisthesis, and lumbar intervertebral disc herniation was limited to a combination of discectomy, decompression laminectomy, and/or fusion with or without instrumentation.22, 23 Recently, the Maine Lumbar Spine Study reported outcomes that indicate only 63% of the patients who underwent a decompressive laminectomy for spinal stenosis were satisfied with the amount of pain after surgery at 4-year follow-up.24 Surgery relieved patients’ pain right away but their pain relief decreased over 4 years. Recent studies have statistically demonstrated that surgery is a superior treatment compared to non-surgical interventions; however, the results of decompression laminectomy and fusion with or without instrumentation demonstrate only fair results in the long term.25–30
One of the historic alternatives for a stand-alone decompressive laminectomy is combining it with an instrumented fusion in order to stabilize the pathological segment. This strategy, however, may result in an increased risk of developing late adjacent segment disease (ASD).31–33 An interlaminar-interspinous implant can be an alternative option in most of these cases. Recent evidence has shown that the investigational device allows flexion, axial rotation, and lateral bending while decreasing the amount of extension,17 versus other interspinous implants that block the amount of extension.8, 9, 34 In comparison to a traditional PLIF, the investigational device reduces adjacent segment accelerated motion and thus reduces the chance of ASD as seen in fusion constructs.21
Recent criticisms of interspinous devices theorize that the disruption of the interspinous ligament could lead to an anomalous ROM. However, Tsai et al demonstrated that destabilized spines with an interlaminar-interspinous implant had similar ROM to intact spines.35
Our retrospective study has demonstrated that the interlaminar-interspinous implant provides long-term pain relief at 2- and 5-year follow-up for patients with low-back pain and leg pain. As previously mentioned, patients with the longest follow-up had the higher pain scores while those with the shortest follow-up had the lowest severity of low-back pain. Although this finding was statistically insignificant, we believe that there is a learning curve for the indications for surgery as well as the surgery itself.
The procedure for placement of the interlaminarinterspinous implant requires less time than fusion and results in lower blood loss. This type of implant could be selected by patients and surgeons who wish to avoid major fusion surgery19, 20 or who have co-morbid conditions that are a contraindication to a standard laminectomy or fusion. The most common indications for implantation in our study were spinal stenosis and/ or lumbar disc herniation. Long-term, our data show that there are very few device-related adverse events and again, at a mean follow-up of 7 years, 98% of the investigational devices were still implanted. Despite the learning curve for proper insertion, the long-term results are promising.
One limitation to this study is that it was a non-randomized, prospective case-controlled study, while some parts of the study were conducted using retrospective data. Further studies need to be done in a randomized, prospective manner comparing the investigational device to an instrumented fusion with respect to long-term clinical outcomes as well as surgery length and blood loss. The coflex interlaminar-interspinous implant is currently undergoing an Investigational Device Exemption (IDE) evaluation and thus is not yet FDA-approved.
EXTENDED REFERENCES
The effects of an interspinous implant on the kinematics of the instrumented and adjacent levels in the lumbar spine.
Lindsey DP, Swanson KE, Fuchs P, Hsu KY, Zucherman JF, Yerby SA.
STUDY DESIGN: Measurement of the kinematics of the lumbar spine after insertion of an interspinous spacer in vitro. OBJECTIVES: To understand the kinematics of the instrumented and adjacent levels due to the insertion of this interspinous implant. SUMMARY OF BACKGROUND DATA: An interspinous spacer (X Stop, SFMT, Concord, California) has been developed to treat neurogenic intermittent claudication by placing the stenotic segment in slight flexion and preventing extension. This restriction of motion by the interspinous implant may affect the kinematics of levels adjacent to the instrumented level. METHODS: Seven lumbar spines (L2-L5) were tested in flexion-extension, lateral bending, and axial rotation. Images were taken during each test to determine the kinematics of each motion segment. The interspinous implant was placed at the L3-L4 level, and the test protocol was repeated. RESULTS: The flexion-extension range of motion was significantly reduced at the instrumented level. Axial rotation and lateral bending ranges of motion were not affected at the instrumented level. The range of motion in flexion-extension, axial rotation, and lateral bending at the adjacent segments was not significantly affected by the implant. CONCLUSIONS: The implant does not significantly alter the kinematics of the motion segments adjacent to the instrumented level.
Are the spines of calf, pig and sheep suitable models for pre-clinical implant tests?
Kettler A, Liakos L, Haegele B, Wilke HJ.
Pre-clinical in vitro tests are needed to evaluate the biomechanical performance of new spinal implants. For such experiments large animal models are frequently used. Whether these models allow any conclusions concerning the implant's performance in humans is difficult to answer. The aim of the present study was to investigate whether calf, pig or sheep spine specimens may be used to replace human specimens in in vitro flexibility and cyclic loading tests with two different implant types. First, a dynamic and a rigid fixator were tested using six human, six calf, six pig and six sheep thoracolumbar spine specimens. Standard flexibility tests were carried out in a spine tester in flexion/ extension, lateral bending and axial rotation in the intact state, after nucleotomy and after implantation. Then, the Coflex interspinous implant was tested for flexibility and intradiscal pressure using another six human and six calf lumbar spine segments. Loading was carried out as described above in the intact condition, after creation of a defect and after implantation. The fixators were most easily implantable into the calf. Qualitatively, they had similar effects on ROM in all species, however, the degree of stability achieved differed. Especially in axial rotation, the ROM of sheep, pig and calf was partially less than half the human ROM. Similarly, implantation of the Coflex interspinous implant caused the ROM to either increase in both species or to decrease in both of them, however, quantitatively, differences were observed. This was also the case for the intradiscal pressure. In conclusion, animal species, especially the calf, may be used to get a first idea of how a new pedicle screw system or an interspinous implant behaves in in vitro flexibility tests. However, the effects on ROM and intradiscal pressure have to be expected to differ in magnitude between animal and human. Therefore, the last step in pre-clinical implant testing should always be an experiment with human specimens.
One-year outcome evaluation after interspinous implantation for degenerative spinal stenosis with segmental instability.
Kong DS, Kim ES, Eoh W.
The authors hypothesized that the placement of the interspinous implant would show a similar clinical outcome to the posterior lumbar interbody fusion (PLIF) in patients having spinal stenosis with mild segmental instability and that this method would be superior to PLIF without significantly affecting degeneration at the adjacent segments. Forty two adult patients having degenerative spinal stenosis with mild segmental instability who underwent implantation of Coflex (Spine motion, Germany) or PLIF at L4-5 between January 2000 and December 2003 were consecutively selected and studied for one-year clinical outcome. At 12 months after surgery, both groups showed a significant improvement in the visual analogue scale score and Oswestry disability index score for both lower extremity pain and low back pain. However, the range of motion at the upper adjacent segments (L3-4) increased significantly after surgery in the PLIF group, which was not manifested in the Coflex group during the follow-up. The authors assumed that interspinous implantation can be an alternative treatment for the spinal stenosis with segmental instability in selected conditions posing less stress on the superior adjacent level than PLIF.
A biomechanical evaluation of an interspinous device (Coflex) used to stabilize the lumbar spine.
Tsai KJ, Murakami H, Lowery GL, Hutton WC.
A biomechanical study of an interspinous stabilization spinal implant (Coflex) was carried out using eight human lumbar L4/L5 motion segments. Each motion segment was tested in compression, then flexion/extension, then lateral bending, and then axial rotation at five conditions: 1) intact; 2) partial destabilization (by cutting the supraspinous and interspinous ligaments, the ligamentum flavum, the facet capsules, and 50% of the inferior bony facet bilaterally); 3) stabilization with the Coflex device; 4) complete destabilization with total laminectomy; and 5) stabilization with pedicle screws and rods. The most important result is that the motion segment after destabilization and insertion of the Coflex device does not allow significantly more or less motion than the intact specimen in either flexion/extension or axial rotation. Thus the Coflex offers nonrigid fixation and can return a partially destabilized specimen back to the intact condition in terms of motion in flexion/ extension and axial rotation.
SUPPLEMENTARY DATA
Top primary diagnoses (inpatient interspinous process spacer implants (ICD-9 proc code 84.80)
MedPAR inpatient interspinous process spacer implants
Charges and reimbursement for inpatient interspinous process spacer implants in the United States
- Received November 5, 2008.
- Accepted May 13, 2009.
- Copyright SAS - Spine Arthroplasty Society 2009
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Dr. Thomas Errico will receive royalties from Stryker Corp.; owns stock in K2M, Inc. and Stryker Corp.; consults for Stryker Corp. and Fastenetix, LLC; has received research support from Synthes, Inc., Paradigm Spine, LLC, and Stryker Corp.; and has received support of clinical staff or training from Medtronic, DePuy Spine, Synthes, Inc., and Paradigm Spine, LLC.
Dr. Jonathan Kamerlink has received travel expenses from Paradigm Spine, LLC.
Dr. Martin Quirno has received travel expenses from Stryker Corp. and Paradigm Spine, LLC.
Dr. Jacques Samani has received royalties from, owns stock in, has consulted for, has received speaking arrangements from, is on the Scientific Advisory Board for, and has received travel expenses from Paradigm Spine, LLC.
Robert Chomiak is an employee of, owns stock in, and has received travel expenses from Paradigm Spine, LLC.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.