


Abstract
Background Cervical disc arthroplasty (CDA) has emerged as an alternative to anterior cervical discectomy and fusion (ACDF) for the treatment of cervical pathologies. Studies are on-going to assess the long term outcomes of CDA. This study assessed the safety and efficacy of the Prestige® LP Disc at 84-months follow up.
Methods Prospective data from 280 CDA patients with single-level cervical disc disease with radiculopathy or myelopathy were compared with 265 historical control ACDF patients. Clinical and radiographic follow up was completed pre-operatively, intraoperatively, and at intervals up to 84 months.
Results Follow-up rate was 75.9% for CDA and 70.0% for ACDF patients. Statistical improvements (p < 0.001) in Neck Disability Index (NDI), neck/arm pain, and SF-36 were achieved by 1.5 months in both groups and maintained through 84 months. At 84 months, 86.1% of CDA versus 80.1% of ACDF patients achieved NDI success, (≥15-point improvement over baseline). Mean NDI score improvements exceeded 30 points in both groups. SF-36 PCS/MCS mean improvements were 13.1±11.9/8.2±12.3 points for CDA and 10.7±11.8/8.3±13.6 points for ACDF. Neurological success was 92.8% for CDA and 79.7% for ACDF patients. The rate of Overall Success was 74.9% for CDA and 63.2% for ACDF. At 84 months, 17.5% of CDA and 16.6% of ACDF patients had a possibly implant- or implant-surgical procedure-related adverse event. Eighteen (6.4%) CDA and 29 (10.9%) ACDF patients had a second surgery at the index level. In CDA patients, mean angular motion at the target level was maintained at 24 (7.5°) and 84 (6.9°) months. Bridging bone was reported in 5.9%/9.5%/10.2%/13.0% of CDA patients at 24/36/60/84 months. Change in mean preoperative angulation of the adjacent segment above/below the index level was1.06±4.39/1.25±4.06 for CDA and (-0.23)±5.37/1.25±5.07 for ACDF patients. At 84 months, 90.9% of CDA and 85.6% of ACDF patients were satisfied with the results of their treatment.
Conclusions Prestige LP maintained significantly improved clinical outcomes and segmental motion; statistical superiority of CDA was concluded for overall success.
This investigational device exemption study was sponsored by Medtronic Spinal and Biologics, Memphis, TN. Study approved by the Hughston Sports Medicine Center Institutional Review Board on January 7, 2005. Clinical trial registered at clinicaltrials.gov: NCT00667459. All participants signed an informed consent.
- cervical disc arthroplasty
- anterior cervical discectomy and fusion
- artificial cervical disc
- cervical radiculopathy
- cervical myelopathy
- adjacent level disease
Introduction
Seven cervical disc arthroplasty (CDA) devices have now been approved by the U.S. Food and Drug Administration (FDA) for the treatment of symptomatic cervical degenerative disc disease (DDD). Twenty four-month Investigational Device Exemption (IDE) studies have revealed CDA outcomes to be at least comparable to anterior cervical discectomy and fusion (ACDF).1–6 More recently, longer-term evidence has been published for many of these same devices, demonstrating the continued safety and efficacy of CDA for appropriately selected patients.7–11
Decompression of the neural elements and permanent stabilization of the cervical spine through ACDF is an effective treatment for surgical candidates suffering intractable neck pain and/or increasing neurological deficit. Cervical disc arthroplasty has the potential to maintain anatomic disc space height, normal segmental lordosis, and physiologic motion patterns after surgery. Widely reported complications, which gave rise to the initial interest in cervical artificial disc replacement as an alternative to fusion, continue to be a concern worthy of exploration in these and other long-term studies.12–21
This study was undertaken to investigate the long-term safety and efficacy of the Prestige® LP Disc (Medtronic Spinal and Biologics, Memphis, TN). The FDA-approved Prestige LP disc is an unconstrained ball-in-trough, metal-on-metal articulation composed of a titanium ceramic composite (Figure 1). The early 24-month clinical and radiographic outcomes for this device have been reported previously.22 We report the 7-year data from the FDA IDE study in patients undergoing single-level anterior cervical discectomy and disc arthroplasty with the cervical disc implant and compare them with those in patients undergoing single-level ACDF.

The Prestige LP Cervical Disc.
Methods
Study Design
This prospective multicenter study was conducted under an approved FDA IDE. Patients in the original 24-month study (clinicaltrials.gov: NCT00667459) were consented after institutional review board approval and followed in this FDA-regulated study for an additional 5 years.
From January to November 2005, 280 nonrandomized patients were enrolled at 20 investigational sites and received treatment for single-level cervical degenerative disc disease using a low-profile cervical disc arthroplasty device. The safety and efficacy outcomes for these patients were compared with data from the 265 historical control patients from a previous FDA-approved IDE study (IDE #G010188; clinicaltrials.gov: NCT00642876) with identical inclusion-exclusion criteria who underwent anterior cervical discectomy and fusion with allograft bone and an anterior plate, utilizing a similar surgical approach. Patients in the study were evaluated preoperatively, intraoperatively, and at routine postoperative intervals of 1.5, 3, 6, 12, 24, 36, 60 and 84 months. Adverse events and secondary surgical procedures were recorded at each follow-up interval.
Independent Data Review
As with the publication of the 24-month results,22 the study sponsor delivered the entire database of raw data to independent biostatisticians at Vanderbilt University for analysis. Using the FDA-approved methods from the original statistical plan, analysis by the independent team that is presented in this report reached the same statistical conclusions as the study sponsor's analysis. Statisticians for the sponsor used the statistical software SAS (SAS Institute, Cary NC) to generate the summary tables and WinBUGS to conduct the Bayesian analysis. Independent statisticians at Vanderbilt used R software for the summary tables and JAGS to conduct the Bayesian analysis.
Statistical Analysis
Bayesian statistical methods are increasingly evident in the peer-reviewed literature for spinal device trials.1, 6 Bayesian results are “positive” when, for example, the posterior probability of efficacy ≥ 0.95 (in the more commonly used and more familiar Frequentist approach, evidence for efficacy is generally thought to be provided by p ≤ 0.05). For assessing study data, the Bayesian posterior probability of a statement (“Treatment A tends to be better than Treatment B”) is the probability that the statement is true. Furthermore, 95% Bayesian Credible Intervals (BCI) are provided for study parameters of interest. A detailed discussion of the statistical analysis plan is presented in the publication of 24-month outcomes.22 In addition, time-to-event analysis was performed for adverse events and secondary surgery events using a Cox proportional hazards model with adjustment for propensity score.
Clinical Outcome Measures
Patient-reported clinical outcomes were measured using validated instruments including Neck Disability Index (NDI) and the 36-Item Medical Outcomes Study Short-Form Health Survey23 (SF-36); Neck pain and arm pain numeric rating scales (duration 0-10 multiplied by intensity 0-10, adapted in part from McDowell et al.),24 neurological status, patient satisfaction, and work status were also assessed. Patient examinations were conducted preoperatively and immediately after surgery, and self-reported outcomes questionnaires were completed before surgery and at each postoperative follow-up interval.
The primary clinical outcome measure for this study was Overall Success, a composite measure consisting of all of the following conditions: 1) NDI Success, defined as NDI score improvement of at least 15 points; 2) Neurological Success, defined as maintenance or improvement in neurological status; 3) Functional Spinal Unit (FSU) Success, defined as maintenance or improvement in disc height and no evidence of subsidence; 4) no serious adverse event (AE) classified as implant or implant/surgical procedure associated; and 5) no secondary surgical procedure classified as a “failure” (Table 1).
Definition of Overall Success. All of the following were required for a patient's outcome to be considered an Overall Success (Reprinted with permission from Gornet MF, Burkus JK, Shaffrey ME, Argires PJ, Nian H, Harrell FE: Cervical disc arthroplasty with PRESTIGE LP disc versus anterior cervical discectomy and fusion: a prospective, multicenter investigational device exemption study. J Neurosurg Spine. 2015 Jul 31:1-16. [Epub ahead of print] PMID: 26230424.22).
FSU Success was an FDA-mandated component of Overall Success, but the measurement was frequently difficult or impossible to obtain due to visualization hindrances on radiographic images of both investigational and control patients, in which case those patients were excluded from the Overall Success analysis. For this reason, Overall Success results are presented both with and without FSU Success as a component.
Radiographic Assessment
Plain radiographs were obtained preoperatively, intraoperatively, and at 1.5, 3, 6, 12, 24, 36, 60, and 84 months to characterize the radiographic measurement of the investigational device and to assess fusion success in the control group. Two independent radiologists from the core lab were trained and performed radiographic assessment measurements utilizing an orthopaedic reading system and digitized images of plain radiographs. A third independent reviewer adjudicated conflicting findings.
Using vertebral endplate distances to measure disc height was expected to be difficult due to visualization challenges created by the CDA implant, despite being low profile. Formation of a solid fusion mass can also potentially obscure measurement landmarks after ACDF. For those reasons, FSU height was chosen as an alternative method for evaluating the maintenance of disc height or directly determining whether the implant had subsided. FSU Success was concluded when neither the anterior nor posterior measurement had declined by more than 2 mm compared with the 1.5-month postoperative assessment.
Segmental motion at the index and superior/inferior adjacent levels was measured on lateral dynamic flexion-extension radiographs using the Cobb method. Radiographic Success required angular motion at the level of surgery to be >4° but <20° at each postoperative interval.
Device Safety/Adverse Events
In addition to Neurological Success—maintenance or improvement in motor function, sensory function, and reflexes—the nature and frequency of adverse events were compared between study groups. An AE mapping scheme for terms and categories of AEs was proposed by the FDA, based in part on the study sponsor's internal AE process and in part on the MedDRA (Medical Dictionary for Regulatory Activities) coding system. This hybrid places each of 20 classifications of AEs under a broad body system (e.g., neurological, cardiac disorders), incident (e.g., trauma, infection), or other (associated conditions/systems with small numerical incidence). Adverse events were analyzed and characterized by their nature into categories, and graded according to World Health Organization criteria as “nonserious” (Grade 1 or 2) or “serious” (Grade 3 or 4) events. Reported AEs were likewise classified based on their potential relation to the implant and/or the surgical procedure. The AE data submitted to FDA, and used for this publication, were generated by a committee of three independent physicians which adjudicated all AE relationships and their severity. An adverse event that resulted in a second surgical procedure would cause the patient to be classified as a study “failure” with respect to the Overall Success determination if it required a supplemental fixation, implant removal, or a revision.
Patient Demographics
Patients in the nonrandomized investigational group and the historical control group were similar demographically (Table 2). Propensity scores were calculated using logistic regression modeling and included in the outcome models to adjust for possible effects of demographic characteristics or preoperative measures on clinical outcomes. Covariate balance after propensity score adjustment was examined using ANCOVA or logistic regression.
Patient demographic and preoperative characteristics: median, interquartile range, (mean ± standard deviation) or percent (count) (Reprinted with permission from Gornet MF, Burkus JK, Shaffrey ME, Argires PJ, Nian H, Harrell FE: Cervical disc arthroplasty with PRESTIGE LP disc versus anterior cervical discectomy and fusion: a prospective, multicenter investigational device exemption study. J Neurosurg Spine. 2015 Jul 31:1-16. [Epub ahead of print] PMID: 26230424.22).
Eligible patients, who consented and were enrolled at sites with institutional review board approval, were all considered candidates for single-level ACDF because of symptomatic cervical DDD at a single level from C3-4 to C6-7, including neck and arm pain that was recalcitrant to nonoperative measures. These nonoperative treatment modalities may have included reduction of painful activities, physical therapy, anti-inflammatory medications, and other directed programs, for at least 6 weeks before surgery (Table 3).
Study patient inclusion and exclusion criteria. Reprinted with permission from Gornet MF, Burkus JK, Shaffrey ME, Argires PJ, Nian H, Harrell FE: Cervical disc arthroplasty with PRESTIGE LP disc versus anterior cervical discectomy and fusion: a prospective, multicenter investigational device exemption study. J Neurosurg Spine. 2015 Jul 31:1-16. [Epub ahead of print] PMID: 26230424.22
Results
Patient Accountability
In the investigational CDA group, 280 patients were enrolled and treated at 20 separate sites. The historical control group consisted of 265 patients treated with ACDF. Follow-up rates at 84 months were 75.9% for the investigational group and 70.0% for the control group, based on the availability of Overall Success (without FSU) outcomes. Follow-up rates based on the availability of any data on a patient at a given study period were 82.0% for CDA patients and 76.2% for ACDF patients.
Patient Demographics
Despite the use of an historical control, the investigational and control groups were similar demographically because inclusion and exclusion criteria were the same for both FDA studies. Statistical differences in tobacco use (higher in the control group) and in race were appropriately balanced for statistical comparison after the application of propensity score adjustment techniques. There were no statistical differences between groups with respect to preoperative medical history or condition, medical usage, or pre-operative scores of key efficacy endpoints. Overall, the 2 study groups were similar prior to surgery (Table 2), with potential confounding effects statistically adjusted, such that conclusions reached by the statisticians were based on treatment effect rather than any potential confounding influences.
Surgical Data
More than 90% of patients in each group were treated at the C5-C6 or C6-C7 level using a standard extrapharyngeal anterolateral approach. Mean operative time in the investigational and control groups was 1.49 hours and 1.38 hours, respectively, a statistically significant difference of approximately 6.5 minutes (95% credible interval, 0.01 to 0.21 hours). Blood loss (51.0 mL - investigational and 57.1 mL - control) and hospital stay (0.98 day - investigational and 0.95 day - control) were not statistically different between the groups.
Overall Success
At 84 months, the rate of Overall Success (without FSU height success) in the investigational group exceeded the rate in the control group by 0.109 (95% credible interval 0.011 to 0.208), and with FSU by 0.001(95% credible interval -0.122 to 0.125), respectively (Table 4). The posterior probability that the Overall Success rate (without FSU) in the investigational group was higher than the control group was 0.985, indicating statistical superiority.
Comparisons of efficacy and neurological variables at 84 months.
Efficacy Endpoints
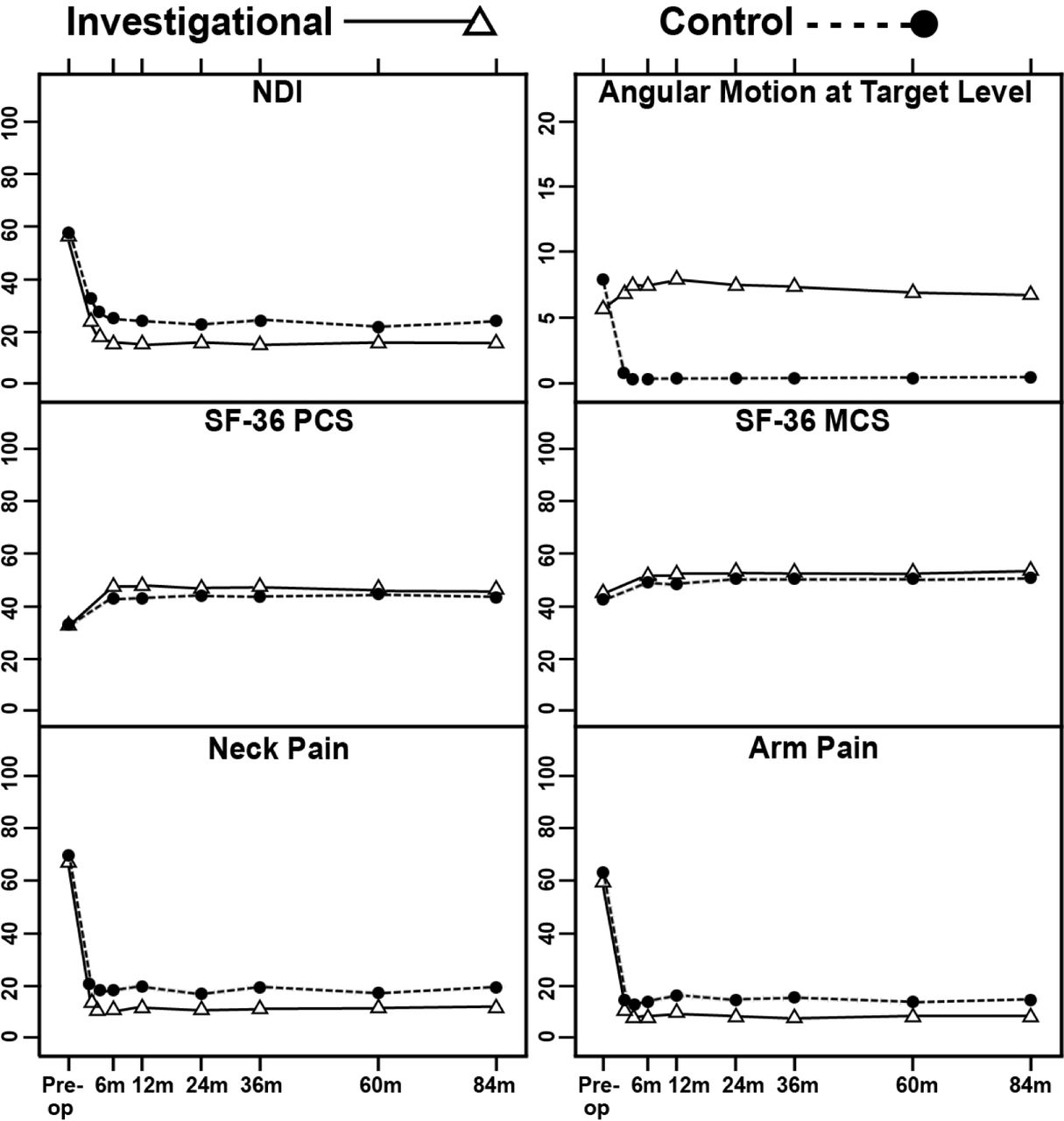
For both the investigational and control groups, improvements in pain and disability scores compared with preoperative assessments were highly significant (p < 0.001, paired t-tests) for NDI, SF-36 PCS and MCS, numeric neck pain and arm pain scores, from 1.5 months after surgery up to 84 months (Figure 2). Mean NDI improvement in both groups exceeded 30 points from 6 months after surgery through 84 months.
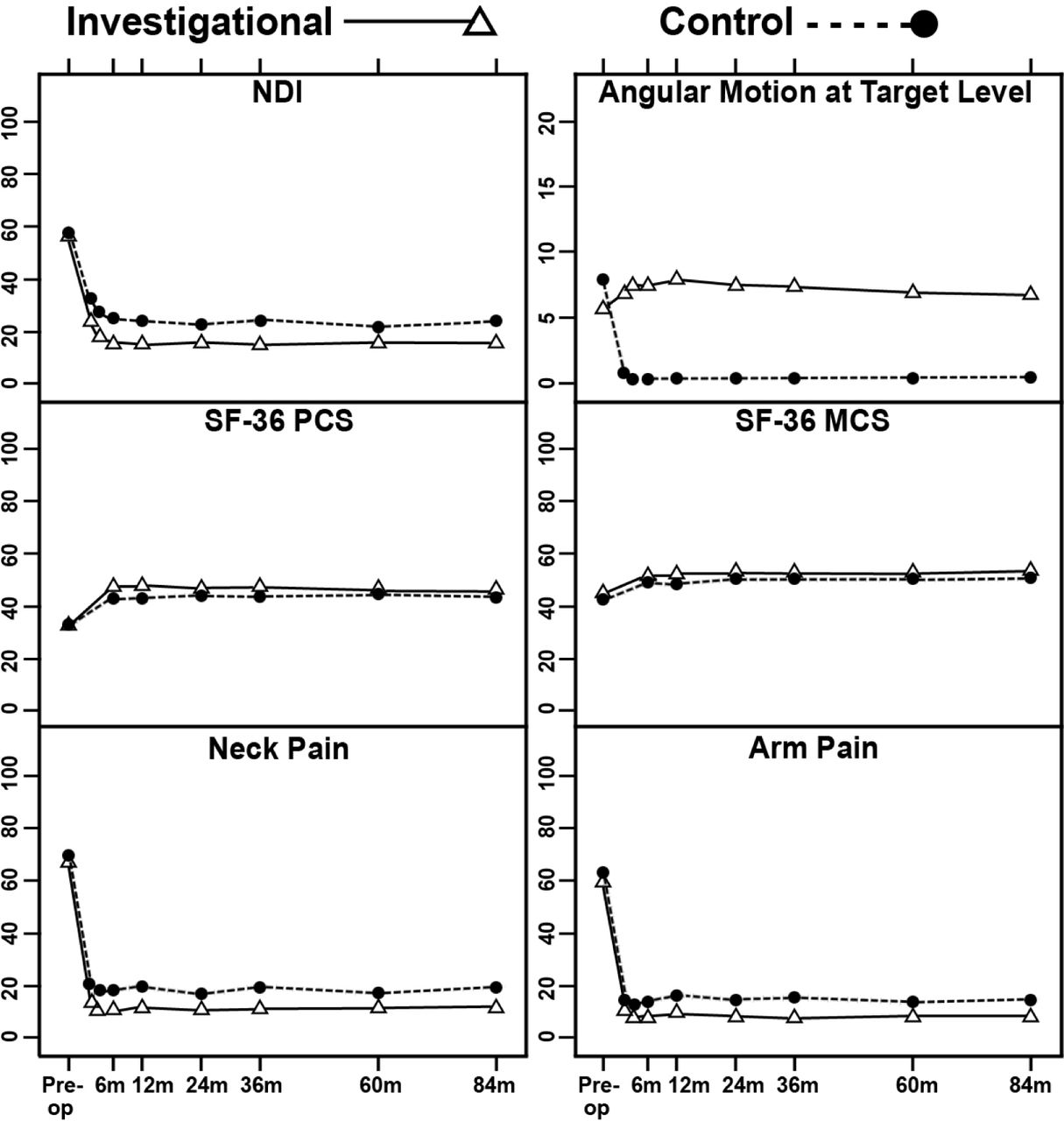
Mean Neck Disability Index (NDI), angular motion at target level, Short-Form-36 Physical Component Summary (SF-36 PCS), Short-Form-36 Mental Component Summary (SF-36 MCS), and neck pain and arm pain scores of the 2 treatment groups at baseline and postoperative time points. Vertical bars indicate 99% confidence intervals of the means.
Bayesian methods were used to evaluate the secondary endpoints. Compared with the control fusion group, the investigational group achieved higher numerical rates of NDI success, neurological success, arm pain success, SF-36 PCS success and MCS success at 84 months; success rates were numerically lower than the control group for FSU height and neck pain (Table 4). The posterior probabilities that the success rate in the control group was less than 10% higher than in the investigational group were essentially 1.0 for all outcomes except FSU height, thus demonstrating statistical noninferiority of the investigational group to the control group for these outcomes 7 years after surgery (Figure 3). The posterior probabilities that the success rate in the investigational group was higher than control group were essentially 1.000, 0.975, and 0.985 for neurological status, SF-36 MCS, and Overall Success without FSU. These Bayesian probabilities exceeded the threshold of 0.95, providing for a conclusion of statistical superiority of the investigational group over the control group for the 3 variables.
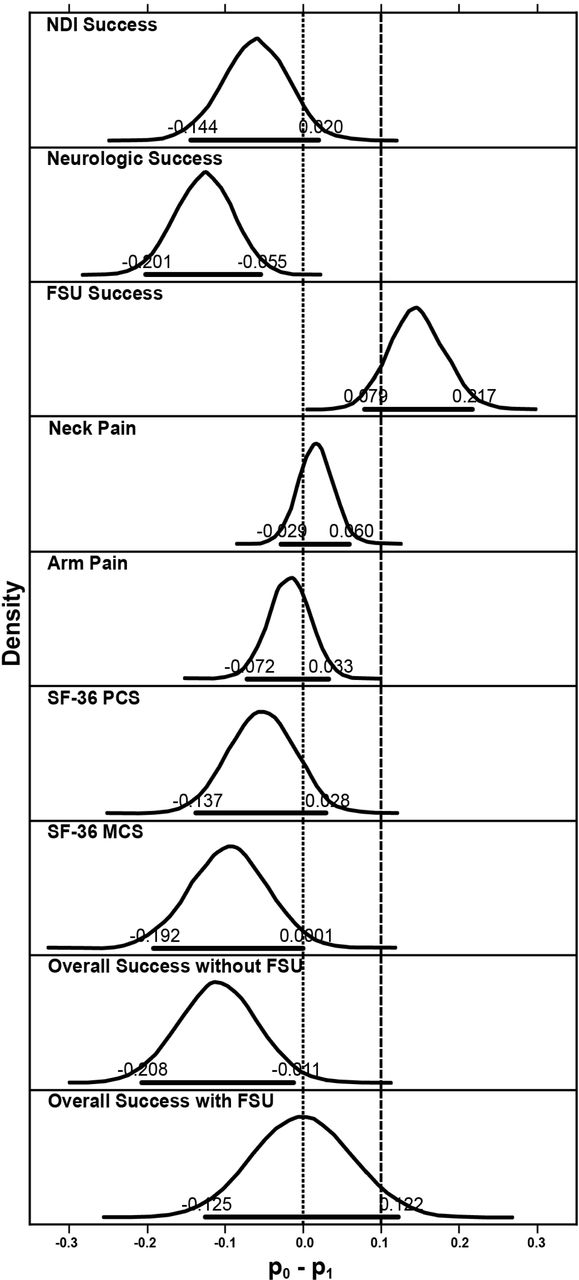
Posterior distributions of differences in success rates of efficacy and neurological variables between the 2 treatment groups. Horizontal bold lines indicate 95% HPD, upper and lower limits of which are also labeled. Dashed vertical line is upper limit of noninferiority and dotted vertical line is upper limit of superiority. FSU = functional spinal unit, HPD = highest posterior density, NDI = Neck Disability Index, SF-36 MCS = Short-Form-36 Mental Component Summary, SF-36 PCS = Short-Form-36 Physical Component Summary.
Treatment Effectiveness/Patient Satisfaction
At each postoperative interval, patients were asked to evaluate the effectiveness of their treatment based on their pain, and in both groups, the percentage of patients choosing “completely recovered” or “much improved” remained consistently high out to 84 months after surgery: 86.1% of investigational and 77.4% of control patients. Seven years after their treatment, approximately 91% of CDA patients and 86% of ACDF patients were satisfied with the results of their surgery and would have the surgery again (Table 5).
Patient satisfaction at 84 months.
Radiographic Assessment
Measurements of the FSU were made by the independent core lab over the course of the 7 years after each surgery in an effort to monitor disc height changes. A success or failure determination could not be made for approximately 40% of the investigational group at 84 months, due to both the gradual reduction in patient follow-up and to the visualization challenges inherent even in this measurement technique, which was a proxy for the more traditional endplate-to-endplate disc height measurement. In those patients for whom a measurement was obtained, FSU Success was above 90% for both treatment groups at all follow-up intervals, with the exception of the investigational group at 84 months with an 83.6% success rate; statistical noninferiority could not be concluded at 84 months.
The core lab also measured angular and translational motion at the index and adjacent levels at each postoperative follow-up visit. In the investigational group, mean angular motion was maintained through 84 months (6.78°), and mean translational motion was consistent throughout the postoperative course at the index level.
Adjacent level motion was also measured preoperatively and at each follow up. In the investigational group, mean angulation at the superior/inferior levels was 8.51°/6.09° preoperatively and 9.31°/6.17° at 84 months. In the control fusion group, superior/inferior adjacent level motion was 10.77°/7.77° preoperatively and 10.71°/8.46° at 84 months. Mean changes in adjacent level angular motion for each follow-up interval are presented in Table 6.
Postoperative mean angular motion at adjacent levels.
Heterotopic ossification was not included in the protocol-defined reporting responsibilities of the independent radiology reviewers. However, bridging bone, defined as evidence of a continuous connection of trabecular bone between vertebral bodies, was reported by the radiology core lab in 13.0% of investigational patients at 84 months. The relatively consistent mean angulation measurements at the index and adjacent levels, and the highly significant mean improvement in pain and disability scores up to 84 months after surgery, indicate little impact clinically from the bridging bone measures.
Adverse Events
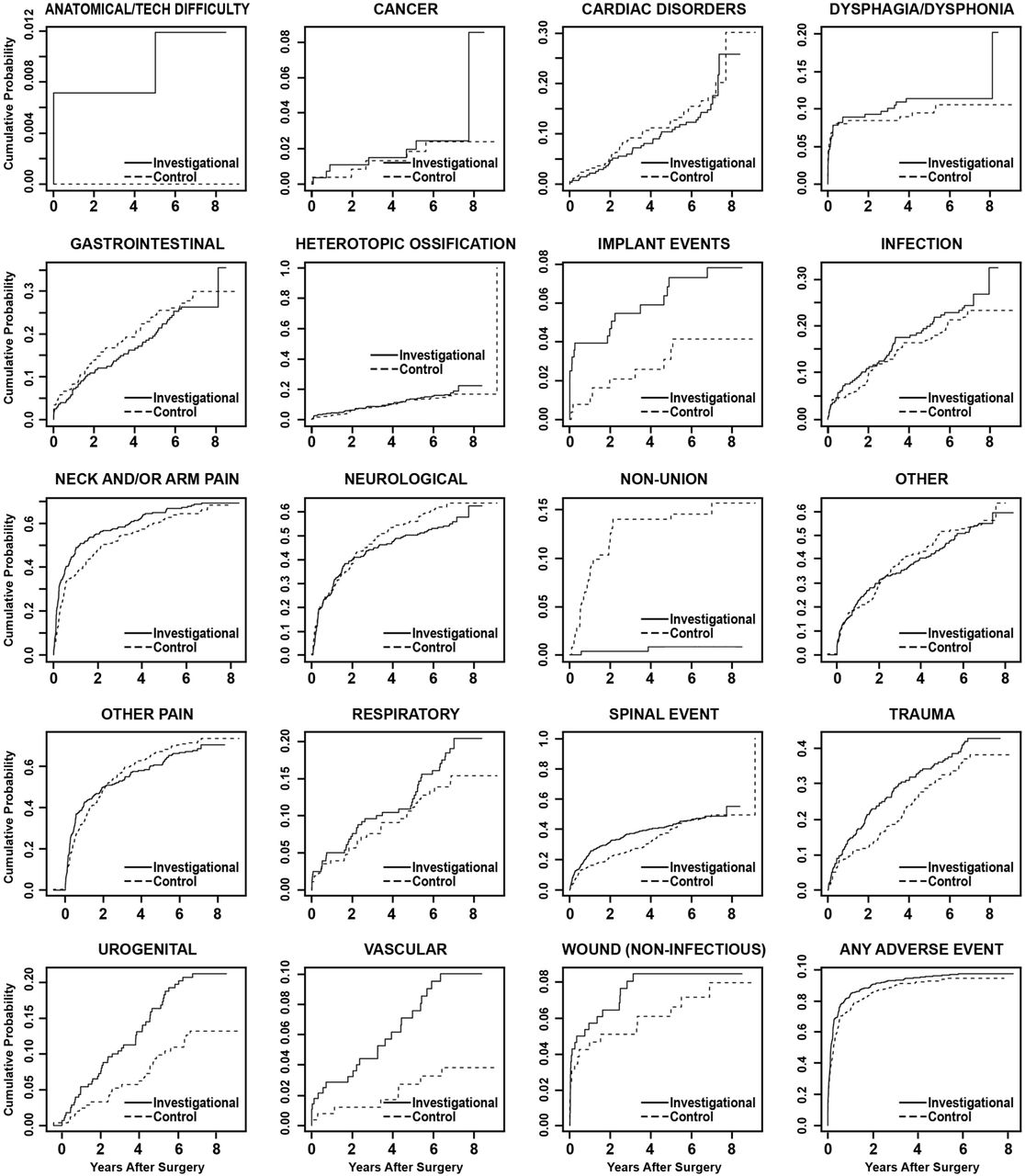
A time course summary of operative and postoperative adverse events is presented in Table 7, reporting the total number of AEs in each category and the number of patients involved. Statistical comparisons of the AE occurrence rates were made using predefined Bayesian methods (Table 8). Up to 84 months, the investigational group had a statistically higher rate of overall AEs, with a higher rate for the categories of anatomical or technical difficulty, implant event, urogenital AE, and vascular AE. As expected, the rate of nonunion events was significantly higher in the control group. To account for patients lost to follow up over the 7-year period, we conducted time-to-event analyses (Figure 4). The conclusion remains similar, except that the category of anatomical or technical difficulty is no longer statistically different between the 2 groups.

Time-to-event analyses (Kaplan-Meier curves) showing cumulative probabilities by adverse event category for the investigational and control groups. P values are based on Cox proportional hazards models, adjusted for propensity score.
Time course summary of adverse events
Comparisons of adverse events at 84-month interval.
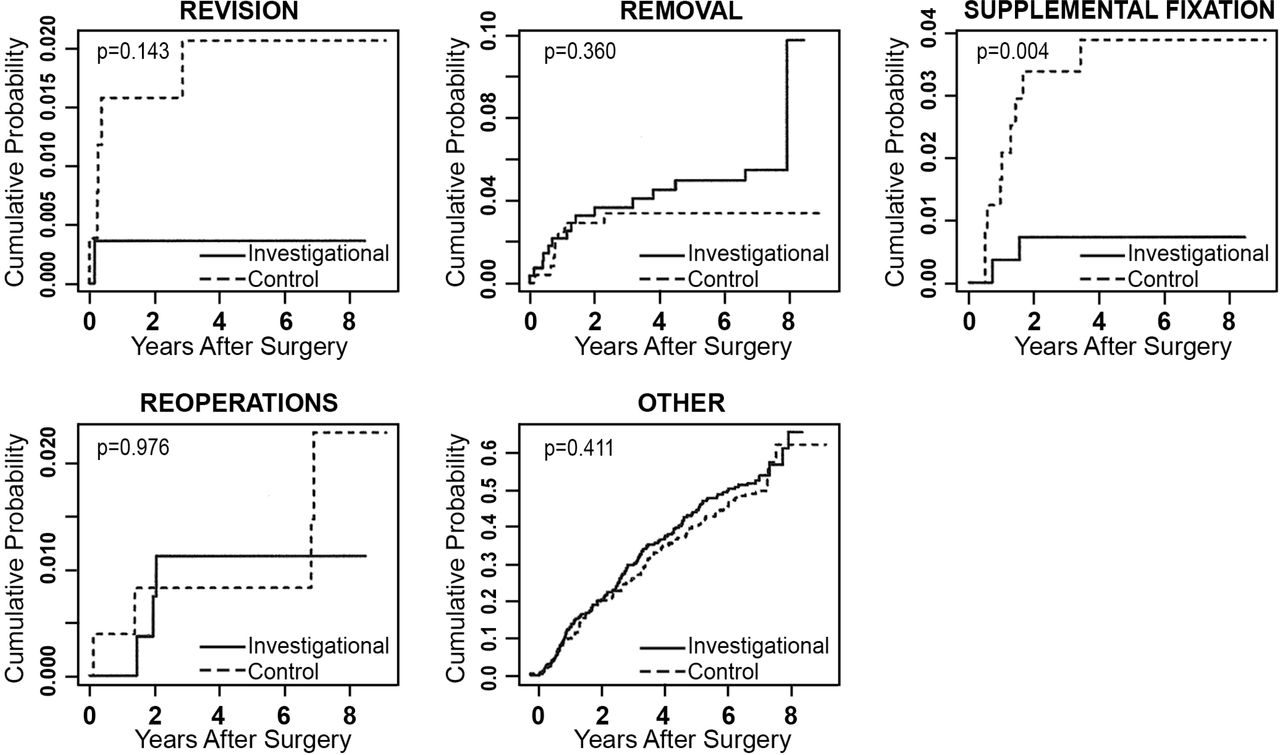
Any adverse event that required a second surgery at the index level was classified according to the protocol in 1 of 4 ways: revisions, removals, supplemental fixations, or reoperations. A revision was defined as a procedure that adjusted or in any way modified the original implant configuration. A removal was defined as a procedure that removed 1 or more components of the original implant configuration without replacement with the same type of device. Supplemental fixation was defined as a procedure in which additional spinal devices not approved as part of the protocol were placed and included supplemental treatments, such as bone growth stimulators. A reoperation was defined as any surgical procedure at the treated spinal level that did not remove, modify, or add any components. Secondary surgical interventions occurred in both treatment groups, and results at 84 months are compared in Table 9. The mean difference in the rate of supplemental fixation procedures between the investigational and the control group was -0.030 (95% credible interval was -0.058 to -0.009). The posterior probability that the investigational group had a lower rate of supplemental fixation procedures was 0.998, demonstrating statistical superiority of the investigational group over the control group. Excluding external bone growth stimulators from supplemental fixation, rates were similar between the 2 groups. To account for patients lost to follow-up, time-to-event analyses were also conducted, with no resulting change in conclusions (Figure 5).
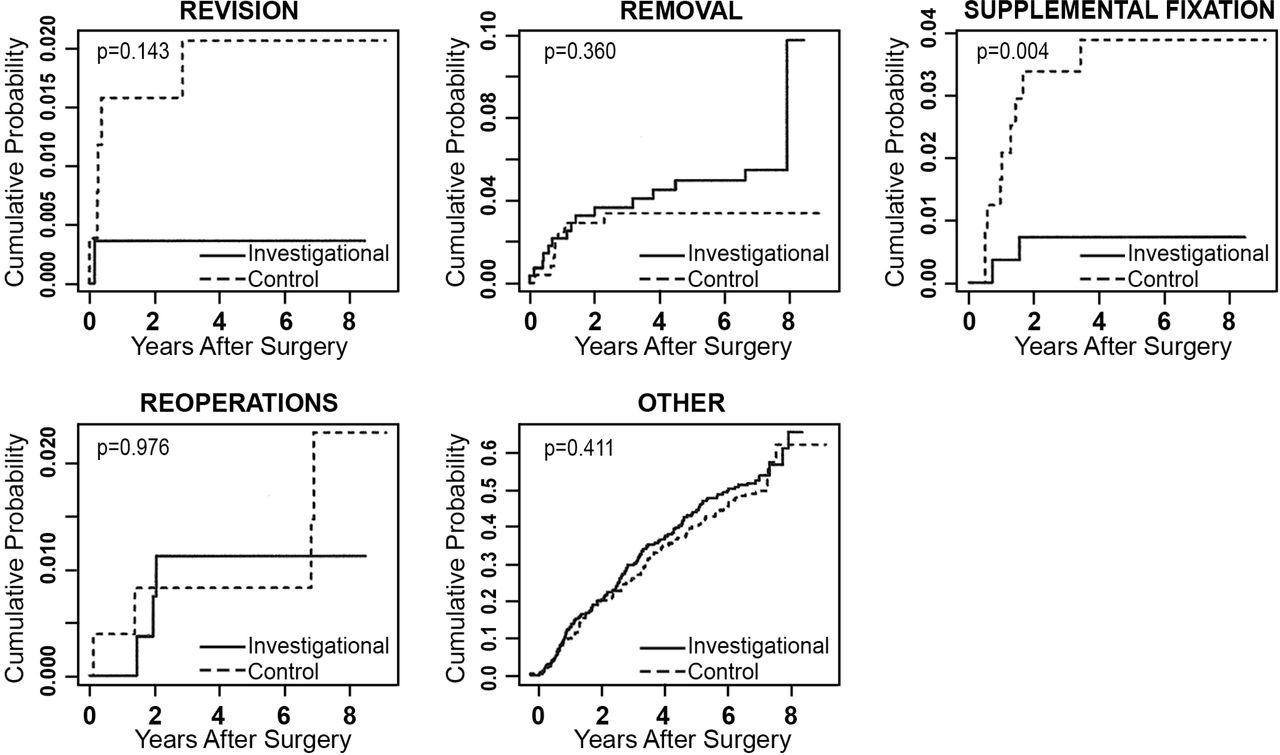
Time-to-event analyses (Kaplan-Meier curves) showing cumulative probabilities of the classifications of secondary surgeries for the investigational and control groups. P values are based on Cox proportional hazards models, adjusted for propensity score.
Comparisons of secondary surgical events at 84 months.
The percentage of patients undergoing secondary surgeries at the adjacent level alone or in conjunction with the index level, cumulatively up to the 84-month follow up, was similar: 9.6% of investigational and 8.3% of control patients.
Device- and device/surgical procedure-related AEs were reported in 17.5% of investigational and 16.6% of control patients. The incidence of AEs that were both serious and classified as device- and device/surgical procedure-related events was also similar in the investigational (6.1%) and control (5.6%) groups.
Discussion
In this study, we report a continuation of the successful results seen at 24 months for patients treated with the Prestige LP Cervical Disc device at 7 years after surgery. Statistical superiority or noninferiority was concluded for CDA compared with ACDF with respect to key clinical and radiographic outcomes, as improvements versus baseline in mean pain and disability measures were statistically significant in both groups at each follow-up interval after surgery. Safety measures including device- and device/surgical procedure-related adverse events and second surgeries were also statistically similar in the investigational and control groups out to seven years. Patient satisfaction was likewise high in both groups.
Articulating prosthetic implants are subject to wear and corrosion following implantation. An advantage of metal-on-metal bearings is the substantially lower volumetric wear debris when compared with conventional metal-on-polyethylene bearing couples. However, implant wear can lead to local and systemic transport of metal debris and increased levels of serum ions. Patients with Prestige LP cervical disc replacements experience increased serum metal ion concentrations after surgery (Gornet MF, Singh V, Schranck FW, Skipor AK, Jacobs JJ: Serum metal ion levels after surgery in patients with metal-on-metal cervical disc arthroplasty. A prospective study up to 84 months. Paper presented at the 30th Annual Meeting of the North American Spine Society. Chicago, IL, October 14-17, 2015). Importantly, the toxicological sequelae of these chronic elevated local and systemic metal levels have not been determined. Several case studies have reported some early local effect of wear debris.25, 26
Cervical disc arthroplasty has the potential for preserving motion at the operated level while providing biomechanical stability and global neck mobility. Restoration or maintenance of physiologic motion at the treated level may result in a reduction in adjacent segment degeneration (ASD) and the need for additional surgery. Although cervical disc replacement and anterior cervical fusion are both safe procedures with a low incidence of significant adverse events related to the procedure, more additional surgeries occurred in the investigational group (CDA) than in the control (ACDF) group. Cervical disc arthroplasty may provide the benefits of neural decompression without placing adjacent motion segments at risk for accelerated degeneration; however, there is no consensus that CDA provides a reduction in ASD rates in longer-term studies.13, 19, 27
The absence of randomization in this study may be considered a shortcoming. This historical control approach, however, was approved by the FDA based on the identical inclusion/exclusion criteria, as well as the Bayesian propensity score techniques which provided statistical balance to all covariates and validated the comparability of the groups.
With one exception,28 the rate of neurological success in the ACDF control group is lower than typically reported.1, 10, 11, 29 We cannot definitively explain this lower rate nor the rate difference between CDA and ACDF in the current study. Caution is advised in making comparisons across studies because measurement and reporting of neurological success varies, therefore, statistical comparisons may not be valid. We can, however, speculate about several possible reasons for the lower success rate in this study: surgical technique, pseudarthrosis rates, ASD, and bias. First, the surgical technique of the CDA involved a more thorough dissection and decompression than that required in the ACDF control group. The CDA required an extensive posterior and posterolateral decompression across the disc space. The entire posterior annulus and posterior longitudinal ligament were resected and uncovertebral joints were partially removed. This decompression may have been less meticulously performed in the ACDF group. Second, relatively high subsidence/pseudarthrosis rates after ACDF have been widely published and may contribute over time to unfavorable changes in neurological status. Third, the increasing incidence of ASD observed after ACDF may be contributing to new neurological pathology--especially as the length of follow-up increases. Finally, we cannot rule out the possible influence of expectations from the treating surgeons. In the absence of blinding, surgeons’ expectations about inferior performance compared with new motion preservation technology may have negatively impacted their assessment of ACDF patients.
Several encouraging clinical and radiographic findings have come from the long-term patient follow-up of this prospective study with concurrent enrollment out to seven years after surgery using the Prestige LP implant. Statistically significant improvements in validated clinical outcome measurements were maintained out to 7 years.
The Prestige LP Disc maintained physiologic segmental motion at 84 months after implantation with a mean flexion-extension difference of 6.78°. Mean translational motion was consistent throughout the postoperative course at the index level. In addition, there were no reported implant migrations. The rates of spontaneous fusion, as measured by the presence of bridging bone, and subsidence, as measured by FSU height, were very low.
Thousands of patients have now undergone cervical disc arthroplasty in very large prospective, multicenter IDE studies in the United States, and this collective body of Level 1 evidence, much of it now with results up to 7 years after surgery, is consistent and convincing in its support of CDA as an alternative to ACDF in appropriately selected patients. In many of these published studies, the outcomes evidence includes a conclusion of statistical superiority for CDA compared with ACDF. Adding to that support is the growing body of longer term cost effectiveness data18, 30–32 that concludes that CDA is economically superior to ACDF. Nevertheless, single-level CDA utilization rates in the United States have thus far trailed expectations due, in part, to limited coverage and reimbursement by insurers.
Conclusions
Patients treated with the Prestige LP Cervical Disc reported significantly improved pain and disability outcomes, at least equivalent to those of the historical control fusion group, 84 months after surgery. Key measures of safety were similar for investigational and control patients. At 84 months, patient satisfaction with the results of surgery was high in both groups. Additional surgical procedures for adjacent segment disease were observed in both treatment groups. Some of the second surgeries involved both index and adjacent levels. Rates for surgery at adjacent levels were similar between the groups and were not statistically significant.
Disclosures & COI
Dr. Burkus reports research support from Medtronic, during the conduct of the study; research support from Medtronic and Zimmer/Biomet outside the submitted work; In addition, Dr. Burkus has a patent with paid royalties from Zimmer/Biomet. Dr. Gornet reports stock ownership in Bonovo; stock in the International Spine and Orthopedic Institute; consultancy for K2M, research support, consultancy, and royalties from Medtronic; stock ownership in Nocimed; stock ownership in OuroBoros; stock ownership in Viscogliosi Brothers Venture Partners, outside the submitted work. Dr. Harrell has nothing to disclose. Dr. Nian reports contract funding for statistical analysis from Medtronic during the conduct of the study. Dr. Schaffrey has nothing to disclose.
This investigational device exemption study was sponsored by Medtronic Spinal and Biologics, Memphis, TN. Study approved by the Hughston Sports Medicine Center Institutional Review Board on January 7, 2005.
- Copyright © 2016 ISASS - This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Multilevel Cervical Disc Arthroplasty: Long-Term Outcomes at 3 and 4 Levels
- Cervical Disc Arthroplasty: Rationale and History
- Adjacent Segment Pathology After Treatment With Cervical Disc Arthroplasty or Anterior Cervical Discectomy and Fusion, Part 1: Radiographic Results at 7-Year Follow-Up
- One-Level Versus 2-Level Treatment With Cervical Disc Arthroplasty or Fusion: Outcomes Up to 7 Years
- Long-Term Clinical Experience with Selectively Constrained SECURE-C Cervical Artificial Disc for 1-Level Cervical Disc Disease: Results from Seven-Year Follow-Up of a Prospective, Randomized, Controlled Investigational Device Exemption Clinical Trial