


ABSTRACT
Background: Diabetes mellitus is one of the leading causes of morbidity resulting in multi-organ dysfunction. Animal studies have shown that hyperglycemia results in stress-induced senescence through the p16-pRb pathway, thereby accelerating early disc degeneration. There is a paucity of literature on the effect of hyperglycemia in human intervertebral disc cells. We aimed to analyze the effect of diabetes mellitus in human intervertebral disc cells.
Methods: This is a prospective study done in patients with degenerative disc disease. Patients were categorized into a control group (no diabetes: 26 patients) and a study group (type 2 diabetes for > 3 years: 24 patients). All patients underwent either discectomy or transforaminal lumbar interbody fusion and the removed disc was transported to pathology department. Tissue was prepared and histopathological grading was done followed by immunohistochemistry studies using antibodies for MMP-1, p21, p16, and pRb.
Results: Samples from diabetic patients had severe (grade 2) degenerative changes compared with the control group (grade 1). Changes were more intense in the nucleus pulposus with increased cellularity and clustering of chondrocytes, and disorganization and loss of nuclear matrix. Immunohistochemical staining for MMP1, p16, and pRb was more intense (Q score = 4) whereas the staining for p21 was less intense (Q score = 1) in the diabetic group compared with the control group.
Conclusion: Our study demonstrates that type 2 diabetes mellitus accelerates stress-induced senescence in human intervertebral discs resulting in early disc degeneration. Also, the severity of disc degeneration is severe compared with the normal subjects.
Clinical Relevance: Hyperglycemia can affect the intervertebral discs similar to other organs and hence adequate control of blood glucose in diabetics can prevent the disc degeneration, which is the initiator of degeneration cascade in spine.
INTRODUCTION
Diabetes mellitus (DM) is a major public health problem with an estimated incidence of 300 million in 2025.1 There are 2 types of diabetes, type 1 or insulin-dependent DM and type 2 or non–insulin-dependent DM, of which type 2 DM accounts for 90% of the cases. DM can affect any organ system, resulting in complications such as cardiovascular disease, chronic renal failure, retinopathy, neuropathy, etc.2 Various studies have postulated hyperglycemia as an important etiological factor for early disc degeneration.3–6 Moreover, the outcome of surgery in patients with DM undergoing surgery for degenerative disc disease is poor compared with nondiabetics.4,5 This effect is mainly because of excessive production of reactive oxygen species, which mediates cellular apoptosis, senescence, and autophagy, ultimately resulting in cellular and organ damage.7–10 Cellular senescence is of 2 types. It could be replicative senescence mediated by the p53-p21-pRb pathway11,12 wherein the cells reach growth-arrest state after repeated cell division, which is irreversible, or it could be induced prematurely by oxidative stress, DNA damage, and mitogenic stress, which is termed stress-induced senescence and is mediated by the p16-pRb pathway.11,12,13 These senescent cells exhibit altered gene expressions, which results in the secretion of degradative enzymes, inflammatory cytokines, and growth factors, and further results in early degeneration and degradation. A human intervertebral disc is composed of an inner gelatinous nucleus pulposus (NP) and an outer annulus fibrosus. The NP is the remnant of notochordal cells and there is a gradual transition of these notochordal cells into chondrocyte-like cells with advancement of age, which is completed by the second decade of life. Studies have shown that hyperglycemia results in stress-induced senescence by the accumulation of reactive oxygen species, which causes cellular damage and degeneration. Thus, we aimed to analyze the effect of DM in human intervertebral disc cells.
MATERIALS AND METHODS
This is a prospective study done in patients with degenerative disc disease admitted in the Department of Spine Surgery, Sri Ramachandra Medical University, Chennai, Tamil Nadu, India. Patients were divided into 2 groups: group 1 was the control group with 26 patients and group 2 was the study group with 24 patients with type 2 DM for more than 3 years. Patients with hypertension, infections, tumors, and inflammatory arthritis were excluded from the study. All the patients underwent either discectomy or transforaminal interbody fusion wherein the degenerated disc fragments were removed and transported to the pathology department of the university. Tissue was fixed with formalin and embedded in paraffin. Histopathological slides were cut at 4 to 5 μm and stained by hematoxylin and eosin. The slides were retrieved and analyzed. One representative paraffin block with adequate material was selected for each subject. Immunohistochemistry (IHC) studies were performed using antibodies for MMP-1, p16, p21, and pRb for all the subjects.
Anti-MMP1 antibody (EP1247Y) is a rabbit monoclonal antibody to MMP1. The matrix metalloproteinases (MMP) are a family of peptidase enzymes responsible for the degradation of extracellular matrix components, including collagen, gelatin, fibronectin, laminin, and proteoglycan. MMP-1 has been shown to degrade bone collagens and also plays a role in bone osteoclastic resorption. MMP-1 is downregulated by p53, and abnormality of p53 expression may contribute to joint degradation in rheumatoid arthritis by regulating MMP-1 expression. The positive control used is skin.
Anti-Rb (phospho S795) antibody is a rabbit polyclonal to Rb (phospho S795). The nuclear phosphoprotein encoded by the Rb tumor suppressor gene is present in many cells and may indirectly regulate cell growth by activating the transcription factor ATF-2. Activation of ATF-2 initiates expression of TGF-β2, which in turn inhibits transcription of genes affecting cell growth. The positive control used is skin.
Anti-p21 antibody is a rabbit monoclonal antibody (EPR18021). The p21protein binds to cyclin–cyclin-dependent kinase complexes and inhibits their kinase activity thereby stopping cell cycle progression. Colon carcinoma was used as a positive control.
All the slides were processed with xylene I for 10 minutes, xylene II for 10 minutes, and alcohol I, II, and III for 5 minutes, each followed by a running water wash for 5 minutes. Antigen retrieval in citrate buffer (pH 6.2) was done for 10 minutes at 95°C followed by a running water wash for 5 to 10 minutes to cool the slides. Peroxide blocking was done with hydrogen peroxide for 10 minutes followed by protein blocking for 10 minutes. Tris buffer wash was given for 5 minutes before incubation with primary antibody for overnight at room temperature. They were then washed with Tris buffer I and II for 5 minutes each followed by incubation with secondary antibody conjugated with HRP polymer for two hours at room temperature. After using 3,3′-Diaminobenzidine substrate chromogen for 10 minutes, the samples were counterstained with hematoxylin for 2 minutes. They were then mounted with a cover slip after washing in running water for 2 minutes.
The IHC was done in 3- to 4-μm-thick sections using the streptavidin-biotin technique. After deparaffinization, the antigenic determinant sites were retrieved in citrate buffer with steam for 10 minutes. Endogenous peroxide was blocked with 3% hydrogen peroxide. The sections were probed with primary antibodies pRb, p16, p21, and MMP1 (Santa Cruz Biotechnology) overnight at room temperature and incubated with secondary antibody conjugated with HRP polymer (anti-mouse IgG-HRP, Santa Cruz Biotechnology). The binding of antibody was detected by 3,3′-diaminobenzidine, which forms a brown color. Finally the slides were counterstained with Mayer hematoxylin. Monoclonal antibody detected against the antigen was observed as brown color in the cytoplasm of chondrocytes in MMP-1 and took up the nuclear staining in p16, p21, and pRb.
The staining pattern for MMP-1, p16, p21, and pRb was calculated using the Q score. The extent of the IHC stained area was scaled as 0 for no IHC staining, 1 for < 10%, 2 for 10% to 50%, and 3 for > 50% (Table 1). The score for IHC intensity was also scaled as 0 for no IHC staining, 2 for moderate, and 3 for strong IHC staining (Table 1). The final score used in the analysis was calculated by multiplying the percentage of positive cells (P) by the intensity (I): Q = P × I (total score of 9).
Extent and Intensity of Immunohistochemistry (IHC) staining.
A histology grading was done according to the new histological degenerative classification for intervertebral disc degeneration by Rutges et al,14 which included the changes in the endplate, morphology of annulus fibrosis, boundary of annulus fibrosis and NP, and cellularity of NP and matrix of NP (Table 2).
New histological degeneration classification for intervertebral disc degeneration.
RESULTS
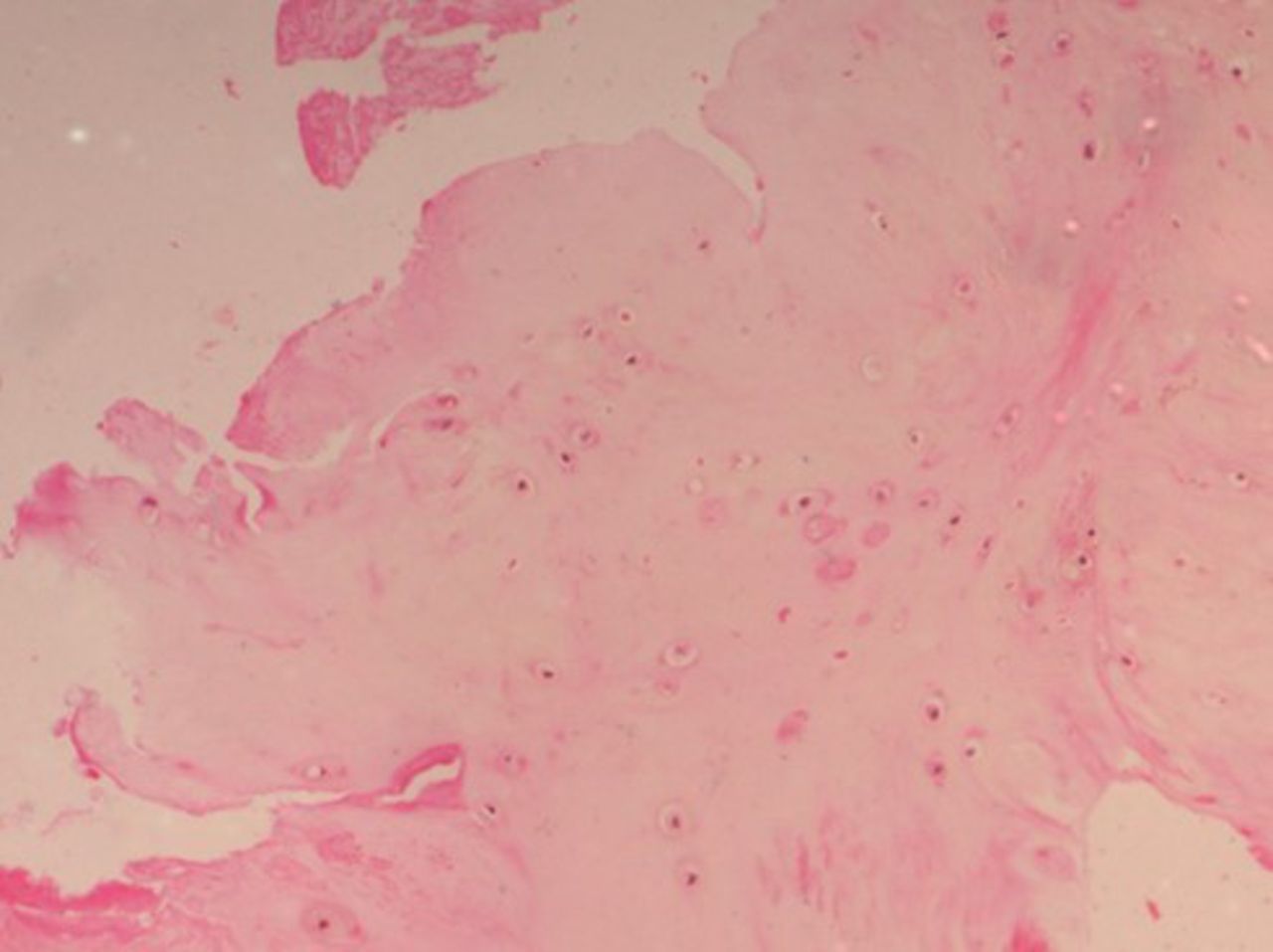
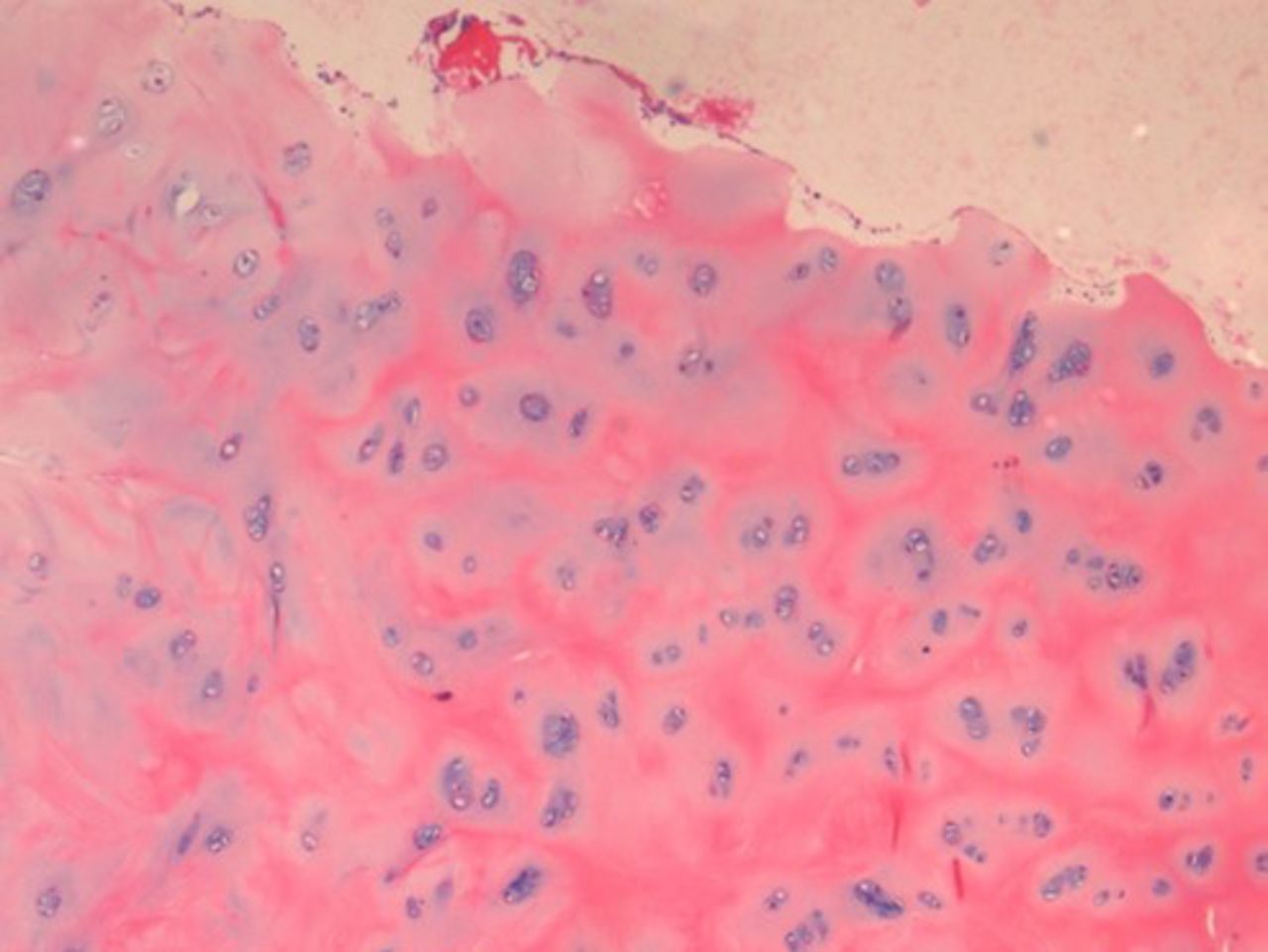
On histopathological examination, the average degenerative score was 8 in the study group compared with 5 in the control group. The changes were more intense in the NP, which showed increased cellularity and clustering of the chondrocytes, disorganization and loss of nuclear matrix, and ruptured annular fibrosis. The severity of degeneration was comparatively less (< grade 1) in the control group (Figure 1) showing mild changes in the NP and annulus as compared with diabetic individuals (Figure 2).
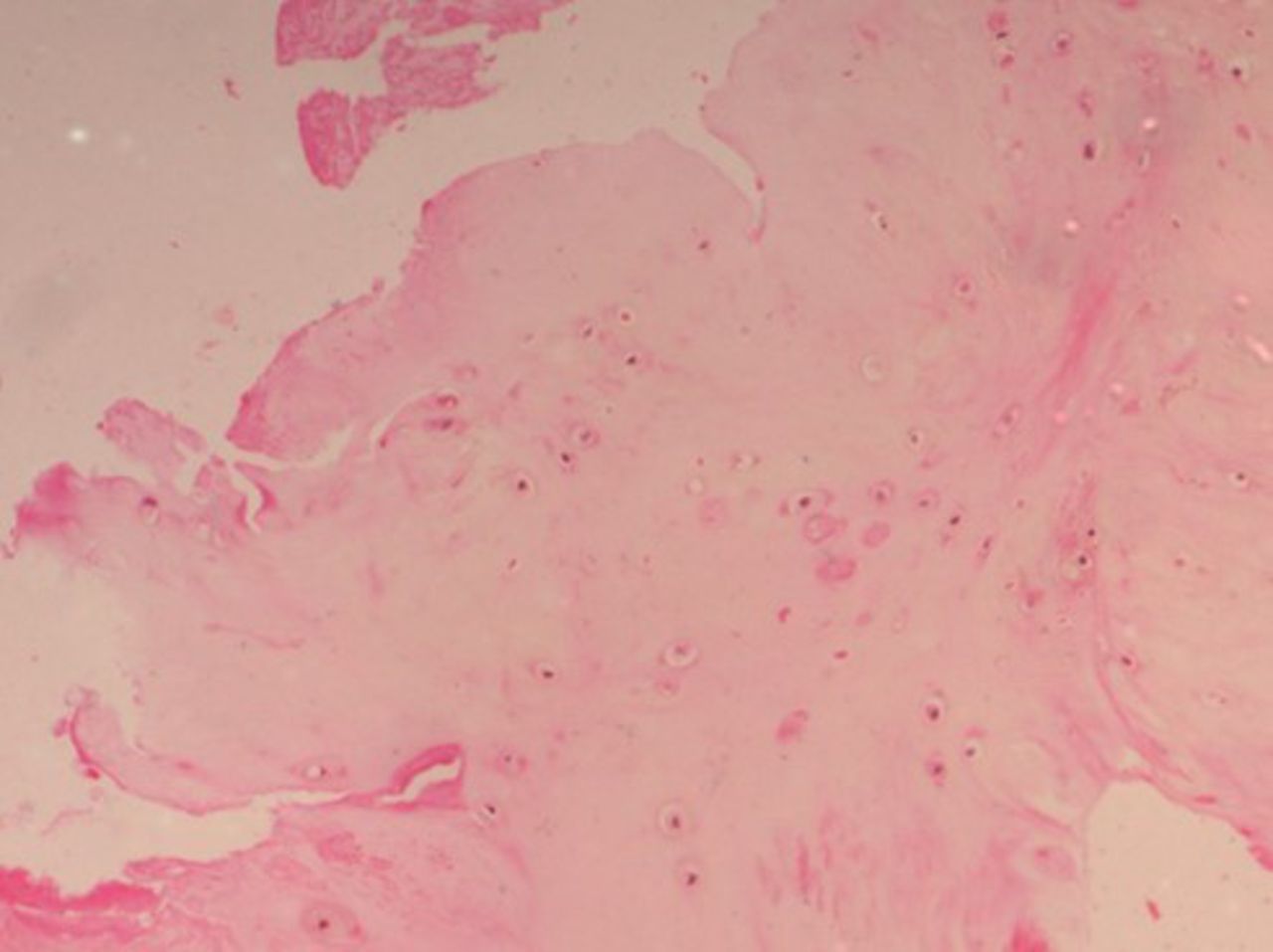
Histopathological examination (HPE) of a control group patient with a lower degenerative score.
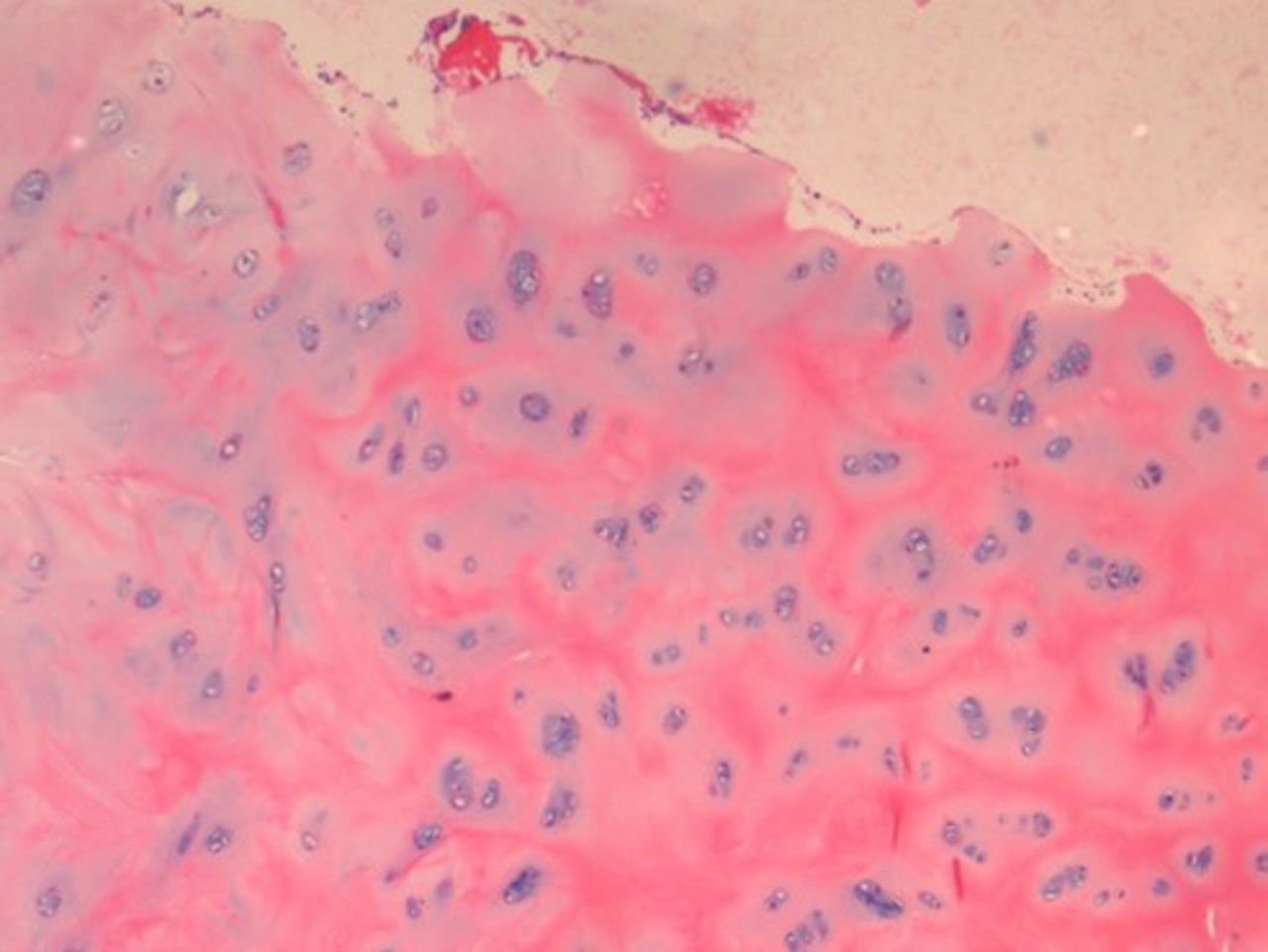
Histopathological examination (HPE) of a diabetic patient with severe degenerative changes.
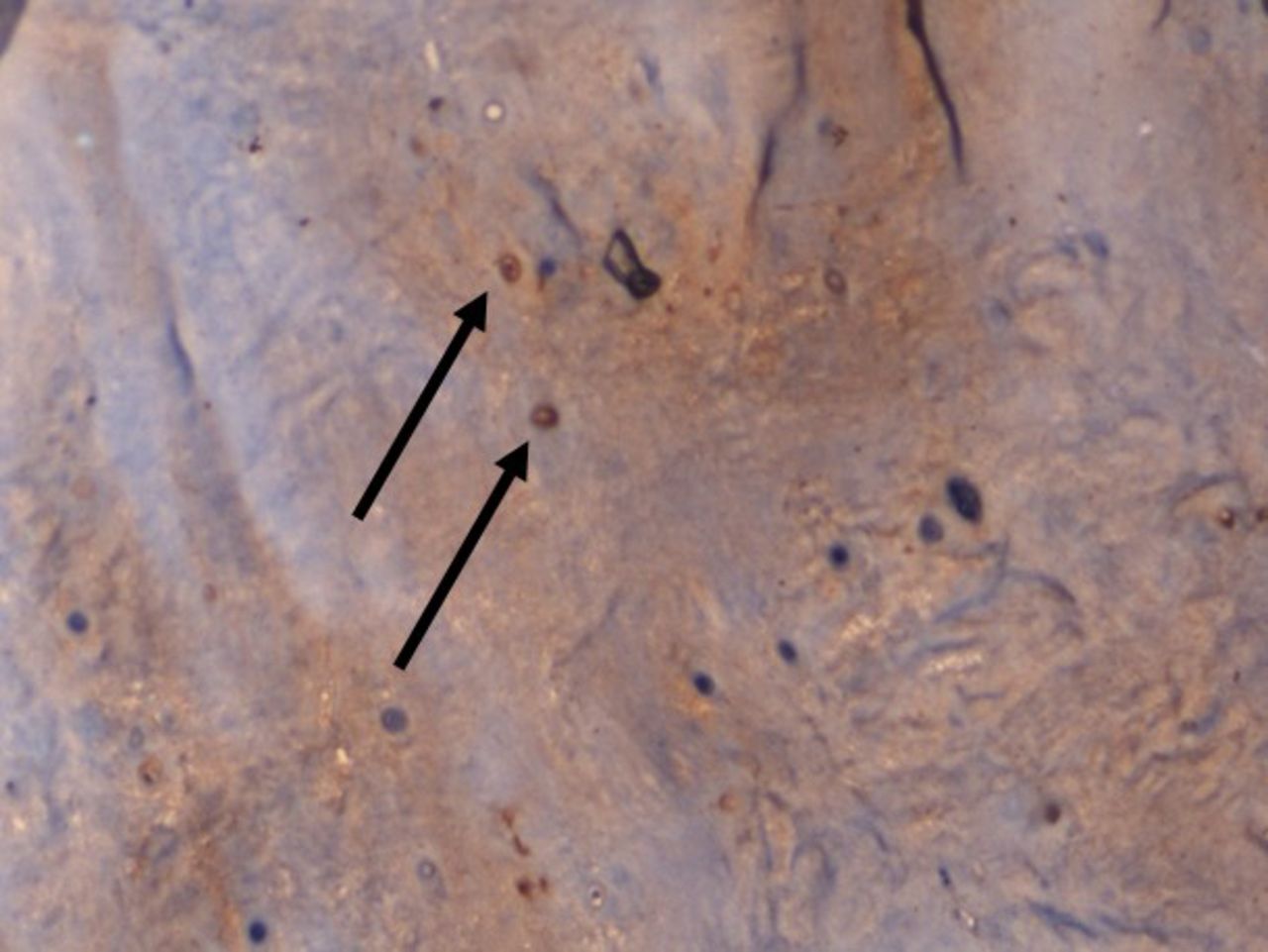
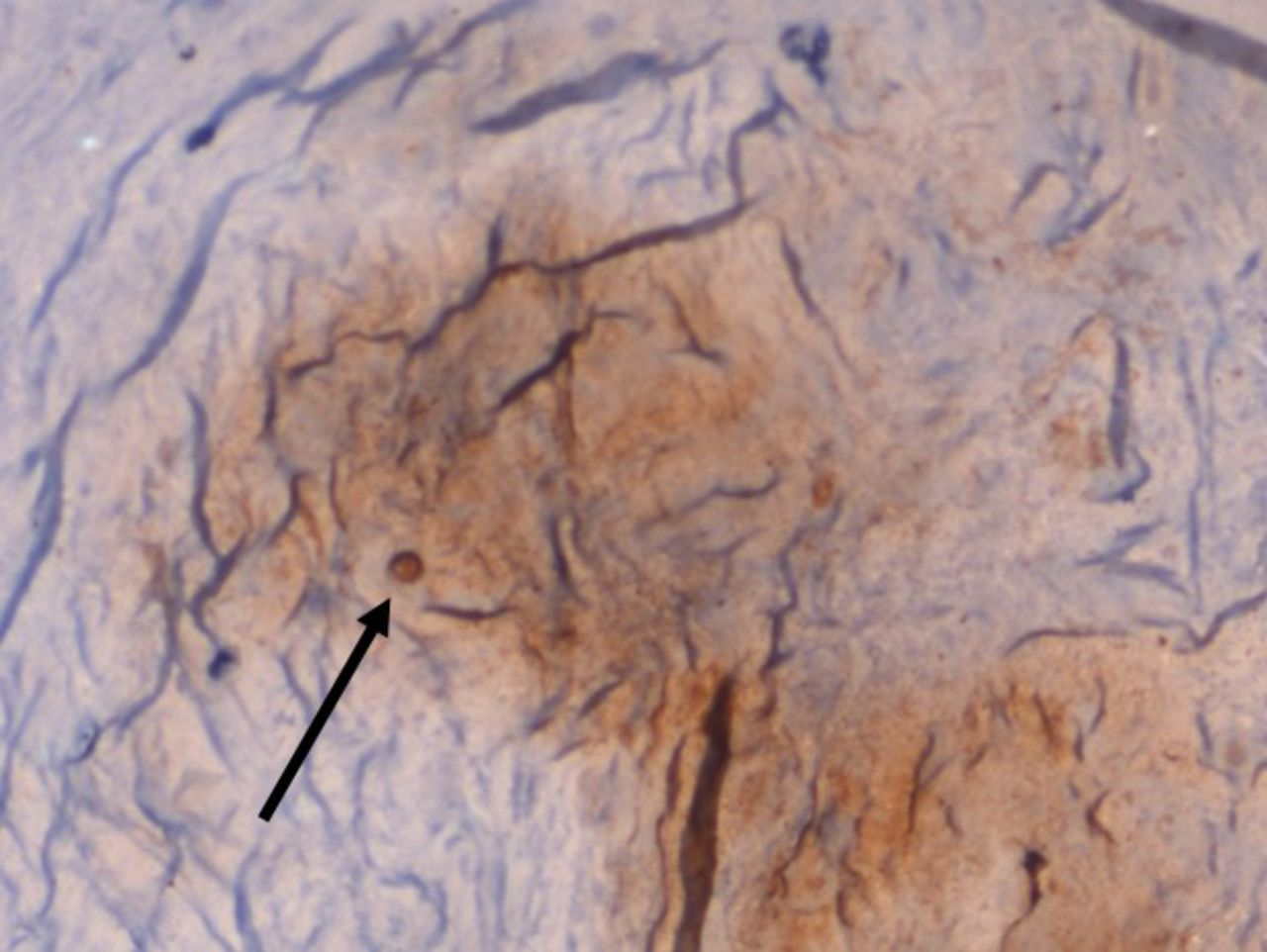
On IHC staining, both the groups showed positive staining for MMP1 with more intense staining in the study group (Q score = 6) (Figure 3) compared with the control group (Q score = 3). The staining pattern of p21 was more significant in the control group (Q score = 6) compared with the study group (Q score = 2) (Figure 4) indicating that the replicative senescence pathway was activated in the control group. The average Q score for p16 was 6 in the study group compared with 1 in the control group; for pRb it was 8 in the study group compared with a score of 3 in the control group (Figure 5). The results indicate that the intervertebral discs undergo stress-induced premature senescence because of hyperglycemia rather than replicative senescence, which occurs in nondiabetic patients.

×100 Immunohistochemistry (IHC) staining for matrix metalloproteinases 1 (MMP1) showing increased cytoplasmic staining pattern in a diabetic patient.
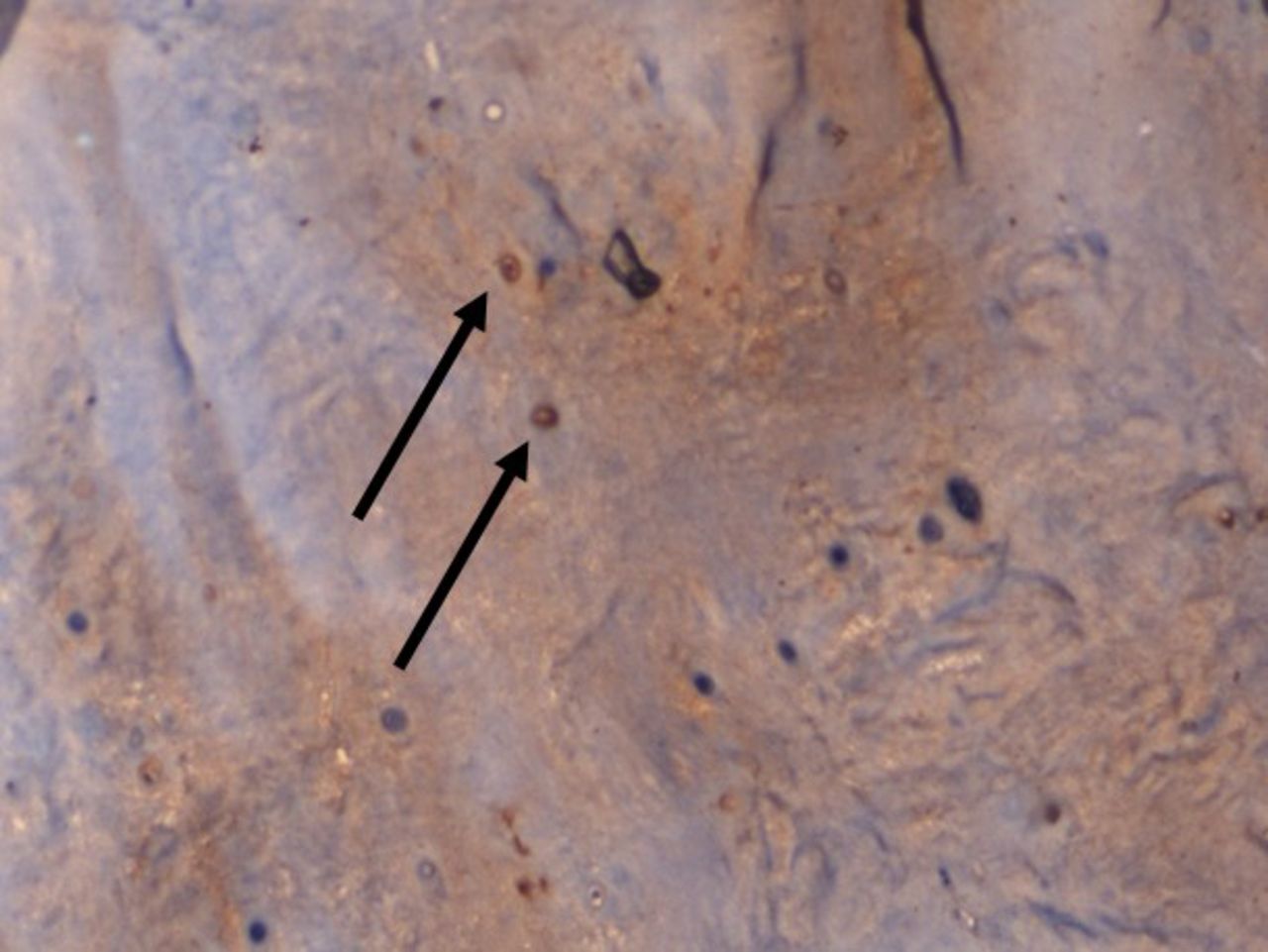
×100 Immunohistochemistry (IHC) staining for p21 showing increased nuclear staining pattern in a nondiabetic patient.
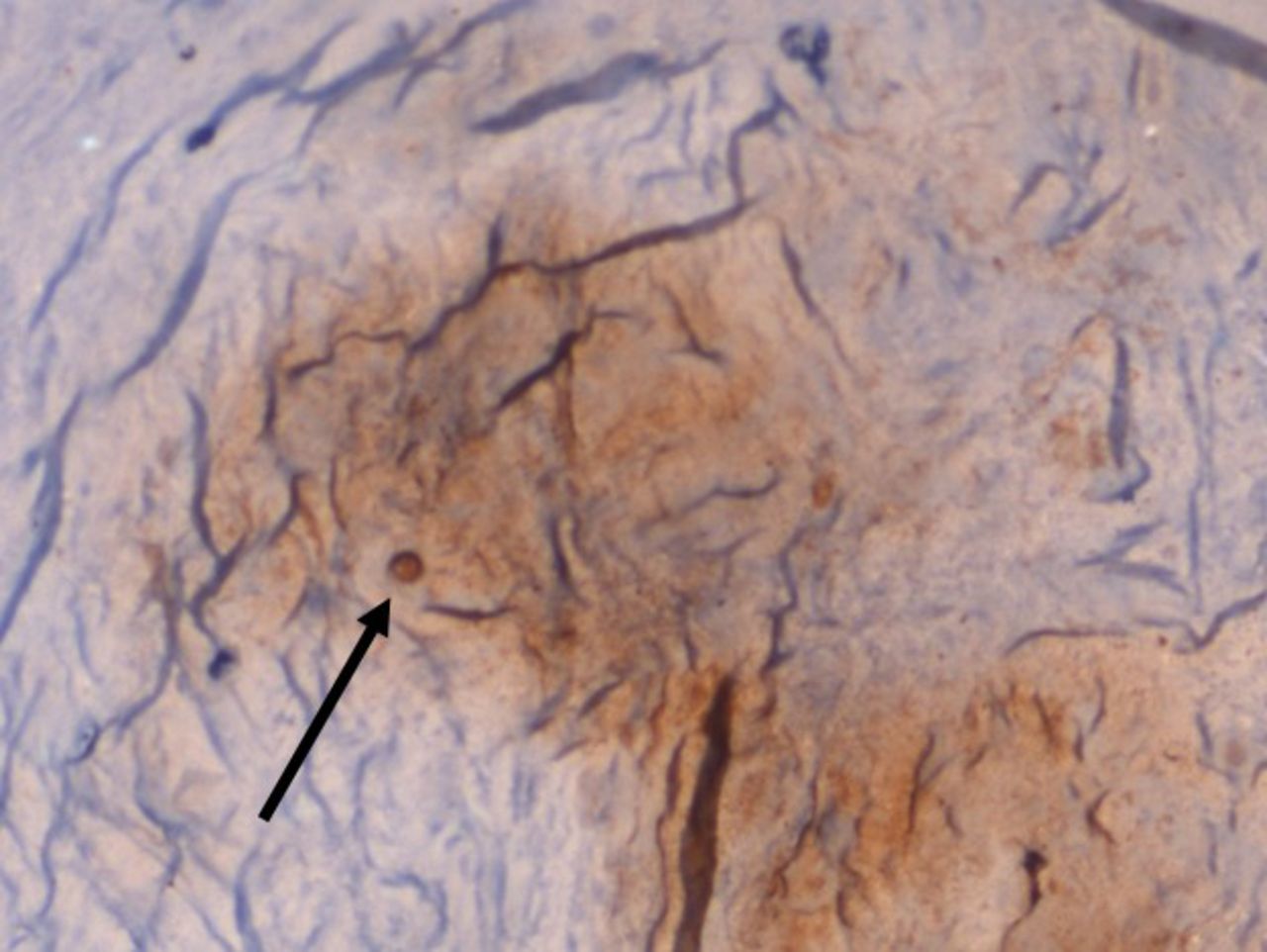
×100 Immunohistochemistry (IHC)staining for pRb showing increased nuclear staining pattern in a diabetic patient.
DISCUSSION
Senescence is a mechanism by which the cells with damaged DNA undergo irreversible cell cycle arrest and are prevented from proliferating, thereby preventing tumor formation. There are 2 types of cellular senescence that have been described. First is replicative senescence mediated by the p53-p21-pRb pathway, which is telomere dependent; the second is stress-induced premature senescence mediated by p16-pRb pathway, which is independent of the telomere length. Animal studies have shown that hyperglycemia accelerates premature disc degeneration by stress-induced senescence. Various mechanisms have been postulated as causes for accelerated degeneration of the intervertebral disc cells, such as increased generation of advanced glycation end products (AGEs) and formation of reactive oxygen species, which in turn results in mitochondrial damage and cellular autophagy.
A hallmark of senescence is increased expression of senescence-associated β galactosidase. However, it is not very specific for senescent cells as cells with high lysosomal content can also stain positive for this assay.15,16 High glucose levels accelerated induced senescence in adult rat NP cells in a dose and time dependent manner rather than replicative senescence as studied by Jae-Gwan Kong.17 Their study showed increased expression of proteins related to p16–pRb pathway (stress-induced senescence) and decreased expression of proteins related to p53–p21 pathway (replicative senescence) in adult rat NP cells treated with both high glucose concentrations. This is similar to the results of our study which showed an increased expression of p16 and pRb and a decreased expression of p21 in the diabetic chondrocytes compared to the control group.
Won et al18 studied the relationship between high glucose and early disc degeneration using diabetic rats with late-onset hyperglycemia. They found premature, excessive apoptosis of NP notochordal cells which significantly increased the MMPs expression in the NP of diabetic rats, leading to disc degeneration and fibrosis. Another study19 confirmed the presence of AGE-producing cells in human degenerated intervertebral discs which resulted in tissue damage and they authors proposed that the increased AGE was due to hyperglycemia. Tsung-Ting Tsai et al20 treated diabetic NP cells from rat coccygeal discs with different concentrations of AGEs for 3 days, and measured the mRNA expressions of MMP-2 and receptor for advanced glycation end plates (RAGE) and also protein expression of MMP-2 activity and extracellular signal-regulated kinase (ERK). There was an increased expression of increase of MMP-2, RAGE, and ERK at both mRNA and protein expression levels in diabetic NP cells. Similar results were obtained in human disc cells obtained from 3 diabetic patients with disc disease.
Various studies21,22 have shown that increased expression of MMPs is responsible for intervertebral disc degeneration and disc herniation. Ho-Yeon Won et al23 demonstrated the increased expression of MMP-1, -2, -3, and -13; TIMP-1; and Fas in diabetic rats at 6 and 12 months of age. They also showed that apoptosis index of notochordal cells was significantly higher in the diabetic rats as was the degree of transition of notochordal NP to fibrocartilaginous NP. They concluded that diabetes is associated with premature, excessive apoptosis of NP notochordal cells which resulted in an accelerated transition of a notochordal NP to a fibrocartilaginous NP leading to early intervertebral disc degeneration. Though in our study, we found an increase in the expression of MMP1 in both the groups, the increase was significantly higher in the diabetic compared with the nondiabetics. We also found that the degeneration was very severe in the diabetic patients as opposed to the control group.
Our study is the first to demonstrate accelerated degeneration and stress-induced senescence in diabetic human intervertebral discs compared with normal subjects. There are a few limitations in our study. We did not evaluate the telomere length or telomerase activity, which signifies cellular senescence. Though the markers in our study represents stress-induced senescence, future studies are required to evaluate the reactive oxygen species and markers of mitochondrial damage in human intervertebral discs.
CONCLUSION
Our study demonstrates that type 2 DM accelerates stress-induced senescence in human intervertebral discs, which can result in early disc degeneration in diabetics. Also, the severity of disc degeneration is severe compared with the normal subjects. Hence, adequate control of blood glucose in diabetic patients can prevent early disc degeneration and prevent the progression of degeneration cascade.
Footnotes
Disclosures and COI: The authors received funding from AO Spine Asia Pacific Research Grant–2017 and report no conflicts of interest.
- ©International Society for the Advancement of Spine Surgery
- This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery. Copyright © 2020 ISASS.
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.