


Abstract
Background Sacroiliac (SI) joint pain is a prevalent, underdiagnosed cause of lower back pain. SI joint fusion can relieve pain and improve quality of life in patients who have failed nonoperative care. To date, no study has concurrently compared surgical and non-surgical treatments for chronic SI joint dysfunction.
Methods We conducted a prospective randomized controlled trial of 148 subjects with SI joint dysfunction due to degenerative sacroiliitis or sacroiliac joint disruptions who were assigned to either minimally invasive SI joint fusion with triangular titanium implants (N=102) or non-surgical management (NSM, n=46). SI joint pain scores, Oswestry Disability Index (ODI), Short-Form 36 (SF-36) and EuroQol-5D (EQ-5D) were collected at baseline and at 1, 3 and 6 months after treatment commencement. Six-month success rates, defined as the proportion of treated subjects with a 20-mm improvement in SI joint pain in the absence of severe device-related or neurologic SI joint-related adverse events or surgical revision, were compared using Bayesian methods.
Results Subjects (mean age 51, 70% women) were highly debilitated at baseline (mean SI joint VAS pain score 82, mean ODI score 62). Six-month follow-up was obtained in 97.3%. By 6 months, success rates were 81.4% in the surgical group vs. 23.9% in the NSM group (difference of 56.6%, 95% posterior credible interval 41.4-70.0%, posterior probability of superiority >0.999). Clinically important (≥15 point) ODI improvement at 6 months occurred in 75% of surgery subjects vs. 27.3% of NSM subjects. At six months, quality of life improved more in the surgery group and satisfaction rates were high. The mean number of adverse events in the first six months was slightly higher in the surgical group compared to the non-surgical group (1.3 vs. 1.0 events per subject, p=0.1857).
Conclusions Six-month follow-up from this level 1 study showed that minimally invasive SI joint fusion using triangular titanium implants was more effective than non-surgical management in relieving pain, improving function and improving quality of life in patients with SI joint dysfunction due to degenerative sacroiliitis or SI joint disruptions.
Clinical relevance Minimally invasive SI joint fusion is an acceptable option for patients with chronic SI joint dysfunction due to degenerative sacroiliitis and sacroiliac joint disruptions unresponsive to non-surgical treatments.
- Minimally invasive surgery
- sacroiliac joint
- sacroiliac joint dysfunction
- sacroiliac joint arthrodesis
- minimally invasive spine surgery
- randomized controlled trial
Background
The sacroiliac (SI) joint was initially identified as a potential pain generator in the early 1900s1 but still remains an under-recognized source of pain localizing to the lower back and buttock.2 The SI joint contains mechanoreceptors3 as well as nociceptive fibers and receptors,4 providing strong evidence that the joint can be a cause of pain. SI joint pathology characteristically gives rise to buttock pain, which may radiate into the lower back, groin, or leg.5 The clinical presentation is often variable and may be mimicked by disorders of the lumbar spine or hip. Blinded studies of local anesthesia to block provoked pain as well as cadaveric nerve root dissections have helped to define the complex innervation of the joint.6, 7 The SI joint requires a combination of articular congruity and balanced muscular/ligamentous compression (i.e. “form and force closure”8) in order to maintain functional stability and facilitate load transfer between the spine and lower extremities. When one or more of these components is compromised, the joint surfaces may be subject to increased stresses resulting in pathologic motion, degeneration and pain. Electromyography has confirmed that, compared to normal patients, those with SI joint pain have altered muscle activation.9
History and physical examination may be suggestive of SI joint-mediated pain, especially when multiple provocative tests that stress the SI joint are found to be positive.10 However, the diagnosis is confirmed when image-guided injection of local anesthetic into the SI joint results in marked pain reduction; a diagnostic approach that has been advocated by numerous practice guidelines.11–15 While radiographic abnormalities of the SI joint are common, they may not reliably correlate with symptoms.16, 17 Multiple investigations have indicated that SI joint-mediated pain is common with a prevalence estimated to be between 15% - 25% among patients with back pain, and even higher following lumbar fusion surgeries.2, 18, 19
Non-surgical treatments for SI joint conditions include medical management, physical therapy (PT), manipulation, steroid injections, prolotherapy, and radiofrequency (RF) ablation. High quality clinical evidence corroborating the benefits of these non-surgical therapeutic options is limited by small patient populations, lack of placebo controls, and failure to utilize validated outcome measures. Blinded controlled trials of intra-articular steroid SI joint injections and RF ablation have demonstrated only short-term20–23 (and in one case long-term24) improvements in SI joint pain.
Arthrodesis of the SI joint was first described in the 1920s25 with several small retrospective case series reporting this operation to be effective for relieving chronic SI joint-mediated pain.26–31 Pain relief is likely mediated by SI joint stabilization, reducing the need for active coordinated musculoligamentous effort to facilitate effective load transfer across the SI joint. However, these open approaches to SI joint fusion typically require long skin incisions with extensive dissection of the soft tissues, subjecting patients to considerable morbidity, pain, substantial blood loss, lengthy hospital stays, prolonged recovery periods, high nonunion rates (9%-41%28, 32, 33), and an inherent risk of serious complications (13.7%34), all of which contribute to wide variation in satisfaction rates (18-80%).34
Minimally invasive arthrodesis of the SI joint has recently been introduced to avoid many of the disadvantages associated with traditional open techniques. The number of these operations being performed increased 4-fold between 2009 and 2012. Currently, they account for over 90% of all SI joint fusions.35 The most widely used system involves the placement of multiple triangular titanium implants coated with a porous titanium plasma spray (iFuse Implant System®, SI-BONE, Inc., San Jose, CA, USA) across the SI joint under fluoroscopic guidance. Several retrospective case series,36–42 a combined multicenter analysis,43 as well as preliminary results from an ongoing prospective multicenter clinical trial (SIFI, NCT01640353),44 have demonstrated that subjects undergoing this procedure exhibited meaningful improvements in pain, disability, and quality of life with high satisfaction rates. Previously published longer-term data demonstrates that this intervention is associated with a high incidence of radiographic fusion across the SI joint with significant pain relief that is maintained at 5-year follow-up.41
To date no published study has concurrently compared surgical and nonoperative treatments for this condition. Herein, we report six-month results of a prospective multicenter randomized controlled trial assessing the safety and efficacy of minimally invasive SI joint fusion relative to staged non-surgical management for patients with symptomatic chronic SI joint dysfunction due to degenerative sacroiliitis or sacroiliac joint disruption.
Methods
Investigation of Sacroiliac Fusion Treatment (INSITE) is an ongoing prospective, open-label multicenter randomized controlled trial. Enrollment took place between January 2013 and May 2014 at 19 spine surgery clinics in the United States. The study protocol (registered on ClinicalTrials.gov [NCT01681004]) was IRB approved at each participating clinical site prior to patient enrollment. The study was sponsored by the device's manufacturer (SI-BONE, Inc., San Jose, CA, USA). All study sites underwent both remote and periodic on-site data monitoring so that all study data were 100% source verified.
Patients were given the opportunity to participate if they were between the ages of 21 and 70 and had a confirmed diagnosis of unilateral or bilateral SI joint dysfunction due to degenerative sacroiliitis and/or sacroiliac joint disruption. Identification of the SI joint as the pain generator was based on a combination of a history of SI joint-localized pain,45 positive provocative testing on at least 3 of 5 established physical examination tests (distraction, compression, FABER test, thigh thrust and Gaenslen's test),10 and at least a 50% decrease in SI joint pain 30 to 60 minutes after image-guided local anesthetic injection into the SI joint performed within 3 months prior to screening. Degenerative sacroiliitis was defined in the study as established SI joint-mediated pain in the context of either radiographic evidence of SI joint degeneration (sclerosis, osteophytes, subchrondral cysts, or vacuum phenomenon) on CT or X-rays or a history of prior lumbar fusion. Sacroiliac joint disruption was defined in the study as SI joint-mediated pain in the context of asymmetric widening of SI joints on CT or X-rays, or the presence of significant contrast leakage during a diagnostic SI joint block. Study inclusion also required the patient to have a baseline score of at least 30% on the Oswestry Disability Index (ODI) and a SI joint pain score (“average SI joint pain in the last week”) of at least 50 on a 0-100 mm visual analog scale (VAS), where 0 represents no pain and 100 represents worst imaginable pain.
Patients were excluded if they met any of the following conditions: inability to diagnose pain related to the SI joint, SI joint pain due to inflammatory conditions, severe back pain deemed to be due primarily to other causes (e.g., lumbar disc degeneration, spinal stenosis, etc.), history of recent (<1 year) major trauma to the pelvis, metabolic bone disease (either induced or idiopathic), or any condition that made treatment with the study devices infeasible or interfered with the ability to participate in physical therapy. Patients involved in litigation, on disability leave, or receiving workers’ compensation related to their back or SI joint pain were also excluded. Patients who agreed to enroll subsequently signed a study-specific informed consent form.
Baseline (pre-randomization) assessments included a detailed medical history, physical examination, and the following quality of life questionnaires, administered by the study coordinator: SI joint and lower back pain using the above-described VAS, ODI,46 EuroQoL-5D (EQ-5D),47 and Short Form-36 (SF-36).48 Oswestry Disability Index (ODI) is a validated ten-question survey for disability due to back pain. EQ-5D is a five-question broad quality of life measure that can be combined into a single index and represents the time trade-off (TTO) utility of current health. EQ-5D also includes a 0-100 mm health thermometer, where 0 means death and 100 means perfect health. SF-36 is a 36-question 8-subscaled generic quality of life measure. SF-36 physical component summary (PCS) summarizes overall physical health, with population norms with mean 50 and standard deviation of 10. Similarly SF-36 mental component summary (MCS) summarizes overall mental health, with similar population norms.
Randomization and Masking
After baseline assessment, subjects were randomly assigned to either SI joint fusion or non-surgical management (NSM). Randomization was stratified by site and underlying condition (degenerative sacroiliitis or sacroiliac joint disruption) in a 2:1 ratio with randomly chosen block sizes of 6 or 9. The 2:1 ratio allowed more precise statistical estimates regarding surgical subject outcomes and promoted enrollment in this patient population seeking relief from chronic SI joint pain. Randomization sequences were computer-generated using a custom algorithm and obtained via a password-protected website after study enrollment. Subjects were not blinded to treatment.
Interventions
NSM, designed to be consistent with current US practices, consisted of pain medications as directed by the site investigator, physical therapy following American Physical Therapy Association (APTA) guidelines,49 intraarticular SI joint steroid injections and radiofrequency (RF) ablation of sacral nerve roots, all of which were delivered in a stepwise fashion and tailored to each individual patient's needs. No cognitive behavioral therapy (CBT) for SI joint pain treatment was used since: 1) no published data supported effectiveness of this method, and 2) standardization of CBT would be challenging, and 3) CBT is not representative of modern healthcare in the US.
Minimally invasive SI joint fusion was performed as described previously36 within 30 days of baseline assessment. All procedures were performed under general anesthesia using fluoroscopy or 3-D computer navigation based on intraoperative CT (O-arm) imaging. A 3-5 cm lateral incision is made over the buttock region and the gluteal fascia is bluntly dissected to reach the outer table of the ilium. A guide pin is passed through the ilium across the SI joint into the center of the sacrum, lateral to the neural foramina. A drill is used to create a pathway into the sacrum and decorticate the bony surfaces. A triangular broach is then passed to prepare the pathway for placement of the implant, which is manually driven into place. In total, two to four implants are placed across the SI joint. The incision is irrigated and the tissue layers are sequentially closed. Subjects requiring treatment of both SI joints could undergo either bilateral same-day surgery or staged procedures. Recorded perioperative measures included estimated blood loss, fluoroscopy time, operating time, number of devices used, and complications. Multiplanar post-placement X-ray or fluoroscopic images of the pelvis were obtained. Subjects were discharged home at the surgeon's discretion; prior to discharge, subjects were re-evaluated for the occurrence of adverse events.
Postoperatively, subjects were asked to remain at heel-toe touch-down weight-bearing using a front-wheeled walker or crutches for three weeks which was progressively increased until fully ambulatory. Beginning 1-3 weeks postoperatively, subjects were asked to undergo individualized physical therapy twice a week for 6 weeks.
Subjects underwent in-clinic follow-up visits at 1, 3 and 6 months postoperatively and study follow-up includes 12, 18 and 24 month visits. Follow-up assessments consisted of a review of adverse changes in health, ambulatory and work status, medication use for pain, physical examination, and quality of life questionnaires. Outcomes of radiographic assessments of subjects assigned to surgery, including pelvic X-ray at 3 and 6 months and high-resolution CT at 24 months, will be reported elsewhere.
Crossover
Subjects were permitted to cross over from NSM to surgery after the 6 month visit was complete. Crossover was required as a design component because investigators believed that: 1) patients with SI joint dysfunction have markedly reduced quality of life (borne out by study data50), and 2) only limited evidence was available to support the effectiveness of non-surgical treatments in this patient population, and 3) promising preliminary results from SI joint fusion using the study device. Investigators also believed that trial enrollment would be severely limited (and potentially not generalizable to standard practice) if the study disallowed crossover in subjects assigned to non-surgical treatment for 24 months. Moreover, because the device was commercially available, patients could have elected to obtain the surgical procedure outside of the study. No subject crossed early and this manuscript focuses on 6-month outcomes only.
Adverse events, defined according to an international clinical trial standard (ISO14155:2011), were monitored continuously and assessed at all study visits. For each event, investigators were asked to rate severity and relationship to the study device, the device placement procedure and, if present, pre-existing conditions. Relatedness was captured as definitely, probably, possibly, unlikely and unrelated to the device, procedure or pre-existing condition. All adverse events were grouped by body system.
Device Description
The iFuse Implant System is a FDA-cleared (K080398) triangular titanium implant that is coated with a porous titanium plasma spray. The triangular shape allows an interference fit that provides immediate stabilization and minimizes micromotion and rotation of the instrumented SI joint. The porous plasma spray coating allows biological fixation of bone, a concept which is commonly utilized by several orthopaedic devices such as hip, knee, and shoulder implants. The iFuse implant is available in configurations ranging from 30-70 mm in length and either 4 or 7 mm in inscribed diameter. The manufacturer recommends placement of at least two implants across the SI joint.
Cohorts, Study Endpoints and Statistical Analysis
The primary analysis cohort consists of subjects who were enrolled (i.e., were eligible and consented) and underwent the assigned study treatment. The primary study endpoint, evaluated at 6 months after the most recent SI joint fusion (to accommodate subjects with staged bilateral surgery), was a binary success/failure composite endpoint. A subject was considered a success if all of the following were met at 6 months: reduction from baseline VAS SI joint pain by at least 20 mm, absence of device-related serious adverse events, absence of neurological worsening related to the sacral spine, and absence of surgical re-intervention (removal, revision, reoperation, or supplemental fixation) for SI joint pain. The 20 mm threshold was selected as the minimum clinically important difference in chronic lower back pain.51, 52 Missing values for the primary endpoint were assumed to be failures. The protocol specified a Bayesian approach to analysis of the primary endpoint; study success was to be declared if the posterior probability that the success rate in the MIS SI joint fusion group exceeded that of the control group was at least 0.975. A non-informative Jeffreys prior distribution was used.53 The study's sample size of 150 subjects was determined to have >80% power under baseline assumptions about success rates (70% for SI joint fusion vs. 30% for NSM). Power calculations assumed no early crossover (none occurred). Full study enrollment was achieved before the first pre-planned interim analysis could be done. Prespecified subgroup analyses included underlying condition (degenerative sacroiliitis vs. sacroiliac joint disruption), history of prior lumbar fusion, smokers vs. non-smokers, and unilateral vs. bilateral SI joint fusion. The study's secondary endpoints included an analysis of patient success rates at other time points as well as improvement from baseline in VAS, ODI, SF-36 PCS and EQ-5D scores. Changes from baseline were compared using t tests or repeated measure analysis of variance (ANOVA). The number of adverse events per subject was compared using Poisson regression and the change in the proportion of subjects using opioids was calculated with a conditional relative odds ratio.54 Confidence intervals for proportions were calculated using standard methods. Analysis of procedure-related variables focused on the index (first side) procedure only. All statistical analyses were performed using R.55
Results
421 subjects were screened for participation, of whom 159 (37.8%) were enrolled. One subject withdrew prior to randomization and 10 withdrew after assignment (7 to surgery, 3 to NSM) but prior to initiation of any SI joint-related treatment, giving rise to a final study population of 148 randomized and treated subjects. All subjects met eligibility criteria with the exceptions of 2 subjects who were over the age of 70 years, 1 subject who had previously been diagnosed with osteoporosis and rheumatoid arthritis, and 1 subject who was currently receiving workers compensation. As the subjects appeared to meet eligibility criteria at study enrollment, their outcomes are included in all analyses.
Baseline Characteristics
Patient characteristics are listed in Table 1. The mean subject age was 51 years and 18 (12.2%) were 65 years of age or older. The majority (94.6%) of subjects were Caucasian and approximately two-thirds were female. Subjects were highly debilitated by SI joint pain as indicated by high baseline pain ratings (mean 82.3 on the 0-100 scale) and ODI scores (mean 61.9). Nineteen percent were not working due to chronic pain. The duration of pain prior to enrollment averaged 6.4 years (range 0.47 - 40.7 years); 87.2% had pain for ≥1 year and 73.6% had pain for ≥2 years. Reported pain locations centered over the posterior superior iliac spine, but distant and/or pain radiating anteriorly or posteriorly was also frequently reported (Figure 1). A large proportion of subjects (37.8%) had a history of prior lumbar fusion, 14.9% had a history of lumbar stenosis, 10.1% had a history of hip disorders, and 7.4% had a history of sacral trauma. SI joint pain persisted despite prior treatments with physical therapy (72.3% of subjects), SI joint steroid injections (85.8%) and RF ablation of the SI joint (16.2%). Approximately two-thirds were taking opioid pain medications at baseline and all reported that multiple activities commonly caused or worsened their SI joint pain. Quality of life (QOL) was substantially diminished, as indicated by low EQ-5D scores (mean of 0.45 on TTO and 55 on health thermometer) and low SF-36 scores (mean PCS of 30.4 and MCS of 43.1), confirming that their SI joint pain represented a significant burden of disease.50
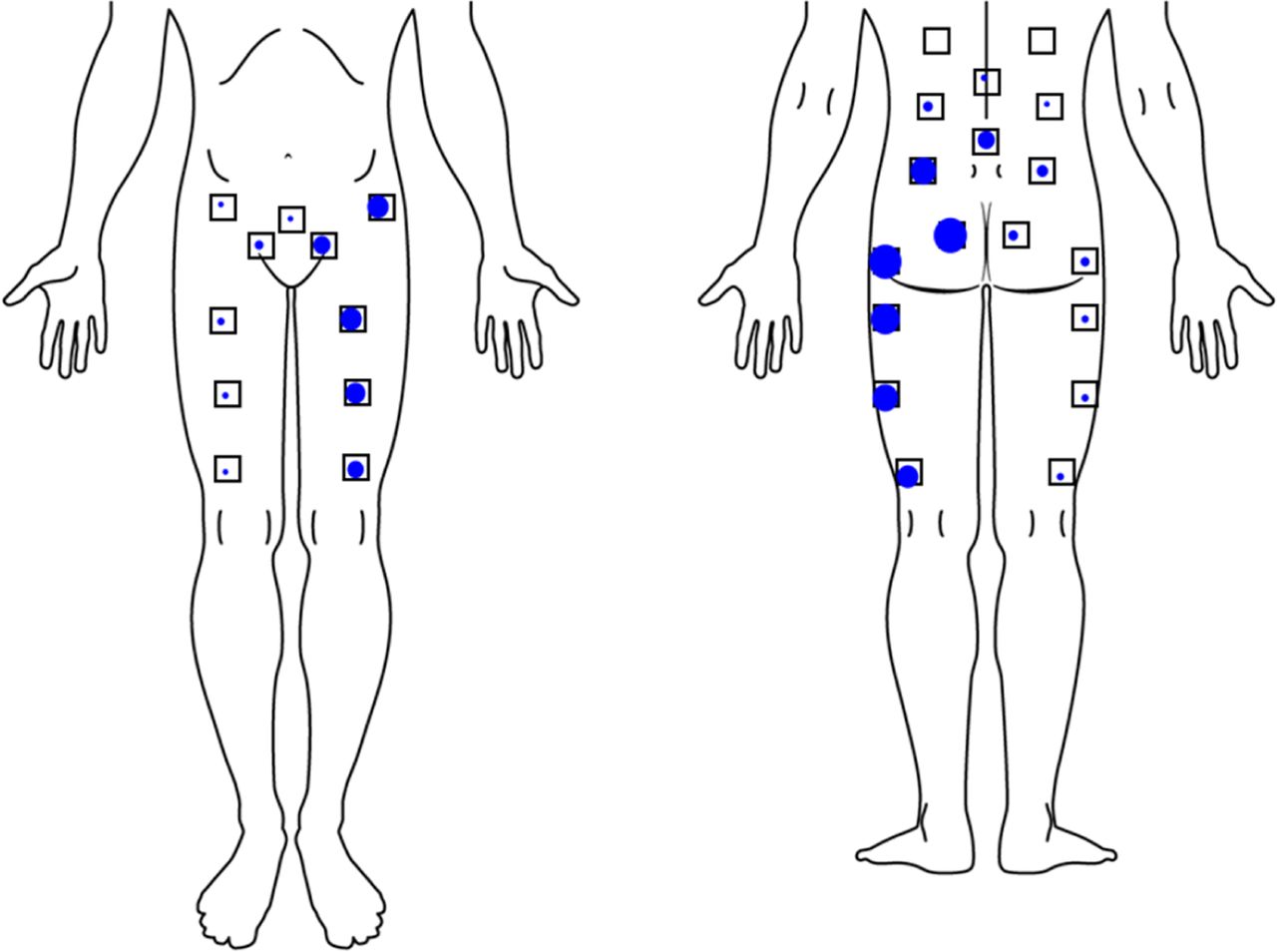
Pain location in subjects reporting primarily left-sided SI joint pain. Dot size is proportional to the number of subjects reporting pain in that location. Only locations shown with small squares were asked.
Characteristics of enrolled subjects.
Procedure Characteristics
All study procedures were performed according to the manufacturer's instructions for use. Most subjects underwent unilateral treatment (Figure 2); 26 (25.5%) underwent same-day or staged bilateral treatment. The mean procedure time was 45 minutes with a range of 14-140 minutes (Table 2) and only 21.6% of procedures lasted over one hour. The mean fluoroscopy time was 2.5 minutes (range 0.13 - 25 minutes) and the mean estimated blood loss was 33 cc (range 0.5-250 cc). In the vast majority of cases (91.2%) three implants were used; 2 and 4 implants were placed in 5 (4.9%) and 4 (3.9%) cases, respectively. In addition, 99.3% of implants placed were 7 mm in diameter. Four procedure-related technical complications occurred (3 cases of sacral deformity either complicating implant delivery or preventing placement of a third device and one case of pin binding) but all cases were successfully completed. The hospital length of stay ranged from 0-7 days (median 1 day). All prolonged hospital stays (≥3 days, 3 cases, 2.9%) were related to patient comorbidities and not procedure-related adverse events.
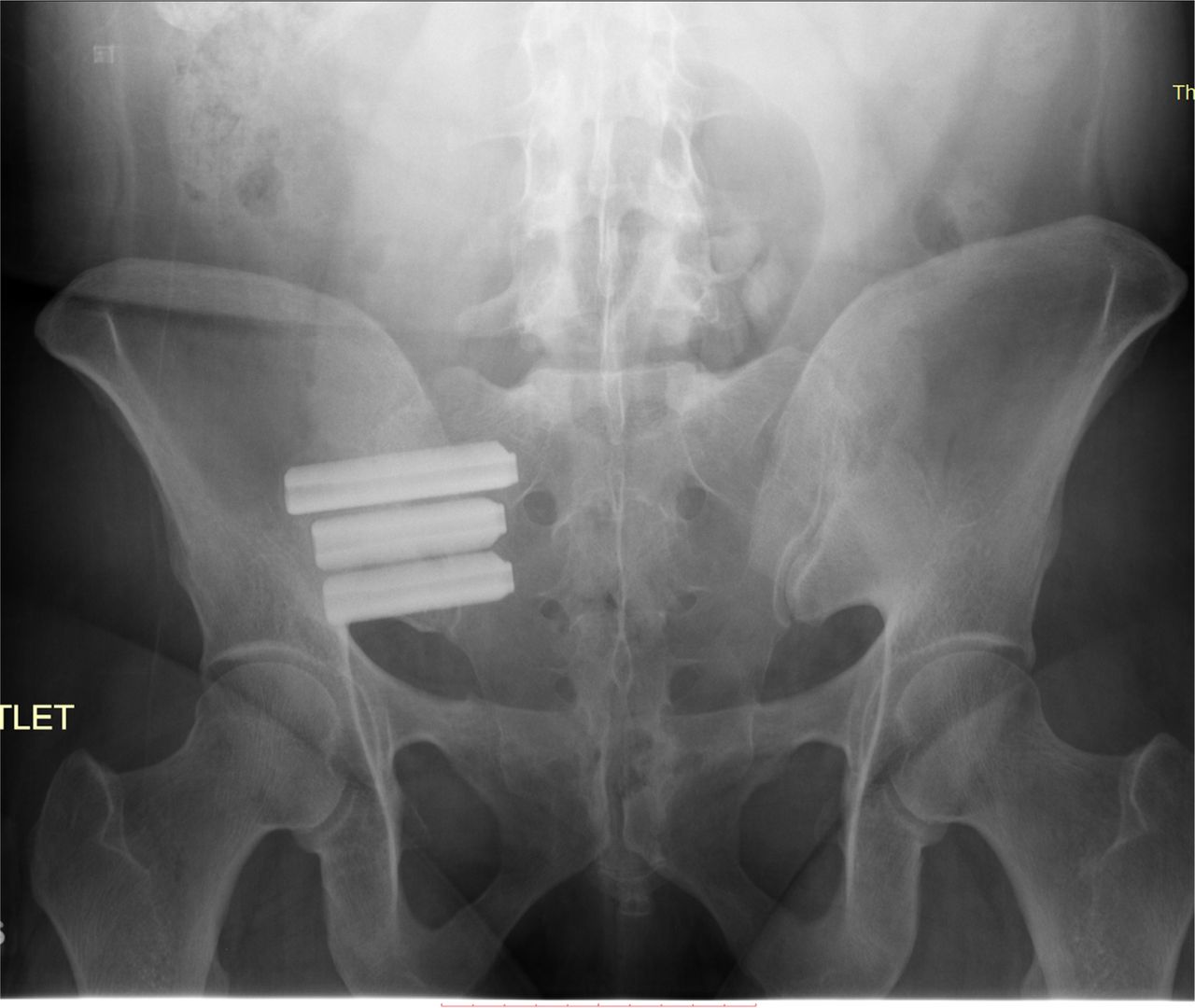
Postoperative outlet view of iFuse devices in the right SI joint.
Minimally invasive SI joint fusion procedure characteristics (n=102). Only the index side procedure is reported.
Non-Surgical Management
Of the 46 subjects assigned to NSM, all but 1 received PT during the six months after treatment assignment. During the first 6 months after assignment to NSM, 34 subjects (73.9%) underwent at least one steroid injection (6 subjects underwent 2 injections) and 21 (45.7%) underwent radiofrequency ablation of the SI joint. Thirty-nine (84.8%) underwent at least 2 types of NSM treatments in addition to pain medications.
Subject Trial Flow
Six-month follow-up was obtained in 44 of 46 (95.7%) NSM subjects and 100 of 102 (98.0%) surgery subjects (Figure 3). Three subjects withdrew voluntary consent and one did not have sufficient follow-up.
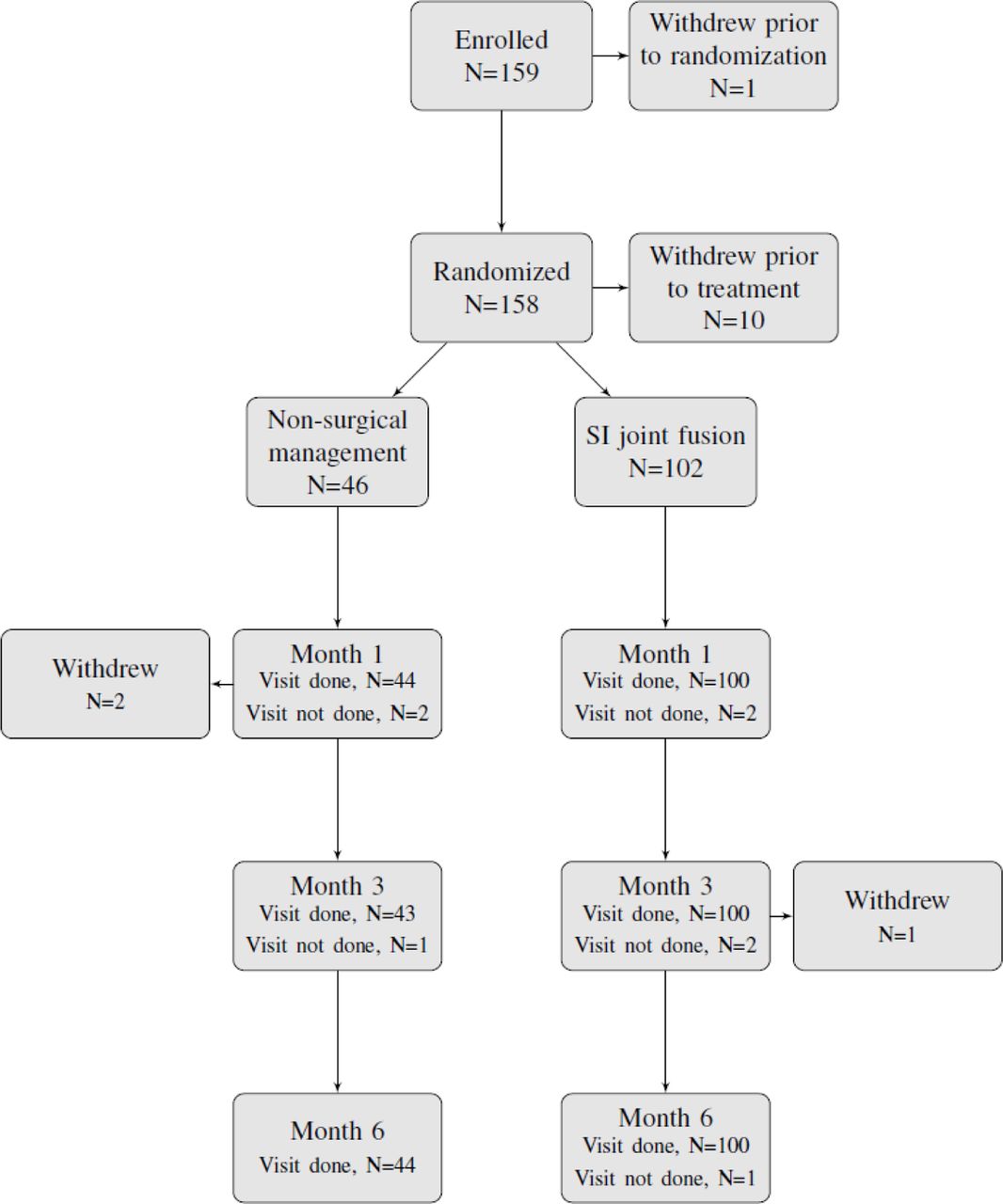
Patient flow.
Primary Endpoint
By month 6, 83 of 102 SI joint fusion subjects (81.4%, 95% posterior credible interval [CI] 72.4-88.4%) and 11 of 46 NSM subjects (23.9%, 95% CI 12.6-38.8%) met the study's primary success endpoint. The difference in success rates was 56.6% (95% CI 41.4-70.0%) and the posterior probability that the success rate was higher in the SI joint fusion group was >0.999. Pre-specified subgroup analysis (Table 3) showed similar differences in success rates by underlying diagnosis, a history of prior lumbar fusion, smoking status or unilateral vs. bilateral SI joint fusion surgery.
Six-month success rates by group.
Pain and Quality of Life Outcomes
In the SI joint fusion group, the mean SI joint pain improved from 82.3 at baseline to 29.8 at 6 month follow-up (a 53-point drop). In contrast, in the NSM group, SI joint pain improved from 82.2 to 70.4 (a 12-point drop, see Table 4 and Figure 4). Changes in SI joint pain from baseline were statistically significant in both groups but the 6-month change was 40.5 points larger in the surgery group (p<.0001). 83% of the surgery group and 25% of the NSM group exhibited a pain improvement of ≥20 points (p<.0001). By 6 months, Oswestry Disability Index (ODI), a measure of disability due to back pain, improved in both groups (mean 30-point improvement in surgery group [p<.0001] vs. 4.9-point improvement in NSM [p=.0524]), but the change was 25 points larger in the surgery group (p<.0001). 75% of the surgery group vs. 27.3% of the NSM group had ODI improvement of ≥15 points (p<.0001). There were no differences in pain improvement or ODI observed across pre-specified subgroups.

Improvement in VAS SI joint pain (top) and Oswestry Disability Index (bottom). Numbers in blue and green show the number of subjects in which the outcome was assessed.
Change in SI joint pain, Oswestry Disability Index, SF-36, and EQ-5D by group and visit.
Quality of life was measured using two generic assessments, EQ-5D and SF-36. The mean EQ-5D time trade-off index which was substantially depressed in both groups consistent with a marked burden of disease,50 improved at 6 months by 0.05 points in the NSM group (p=.1969) and 0.29 in the iFuse group (p<.0001); the mean difference in EQ-5D TTO improvement was 0.23 points (p<.0001).
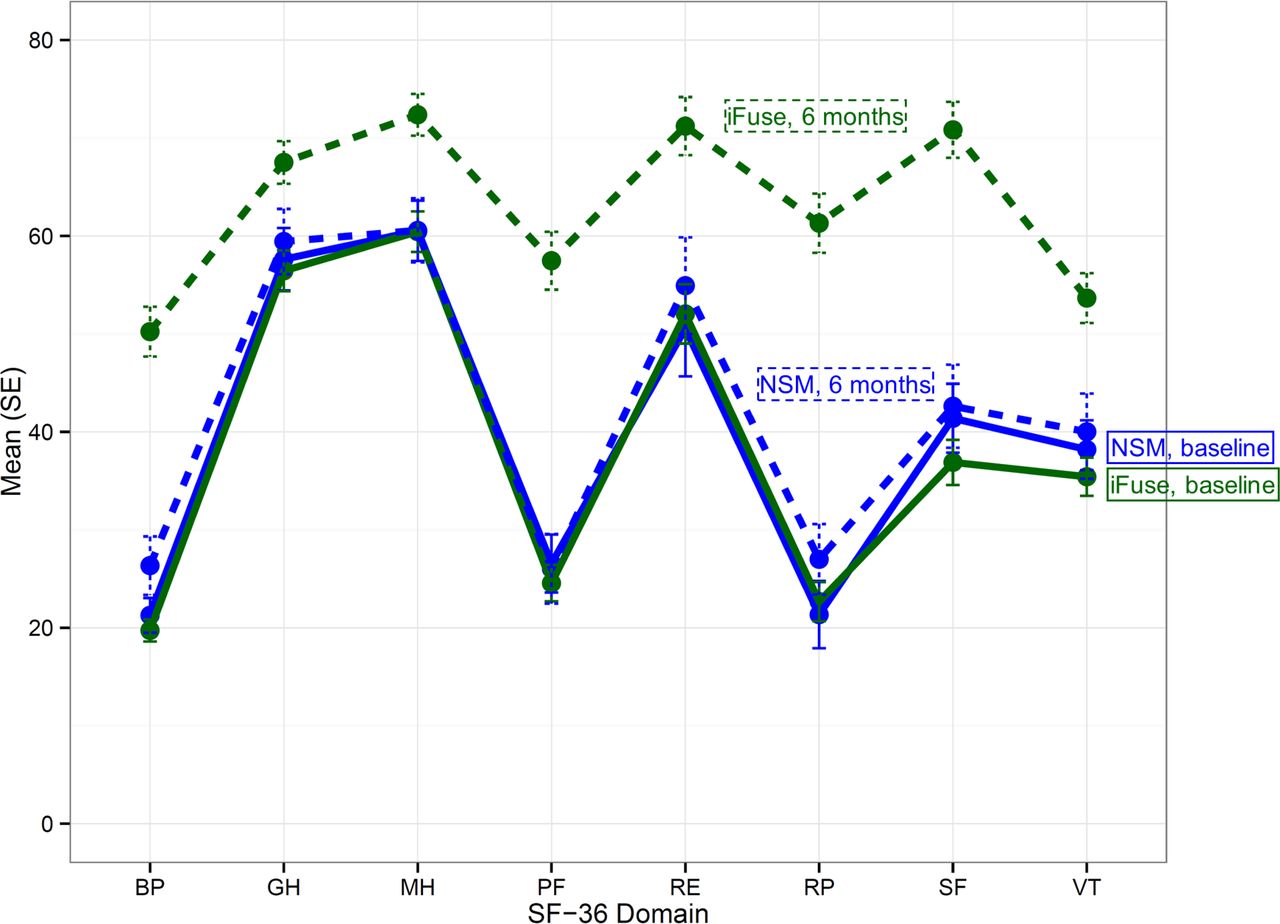
At the present time, the mean ratings for all SF-36 domains improved significantly in the surgery group (p<.0001 each) but not in the NSM group (all p>.05, Figure 5). The mean SF-36 PCS improved by 12.7 points in the surgery group and 1.2 points in the NSM group (p<.0001 for comparison). Similarly, SF-36 MCS improved by 6.2 points in the surgery group and 0.6 points in the NSM group (p=0.0053 for comparison).

Improvement in SF-36 domains at 6 months compared to baseline. Green=surgery, blue=NSM, solid line=baseline, dotted line=6 months. SF-36 domains are BP (bodily pain), GH (general health), MH (mental health), PF (physical function), RE (role emotional), RP (role physical), SF (social function), and VT (vitality).
Six-month satisfaction rates were higher in the surgery group compared to the NSM group (79.0 vs. 27.3% very satisfied, p<.0001); specifically, 81% of surgery subjects said they would definitely have the procedure again. Compared to baseline, opioid use at 6 months decreased from 67.6% to 58% in the surgery group and increased from 63 to 70.5% in the NSM group (p=0.082).
Adverse Events
As of the 6-month follow-up time point, 181 adverse events were reported (133 in the surgery group and 48 in the NSM group). The mean number of events per subject was slightly higher in the surgery group (1.3 vs. 1.0 events, p=0.1857). Leg and pelvic pain were the most common adverse events (Table 5); the likelihood of adverse events in the pelvis was slightly higher in the surgery group (33.3% vs. 21.7%, p=0.1773) and the likelihood of pulmonary events was slightly higher in the NSM group (p=0.0326). Surgical wound problems occurred in 6 (5.9%) subjects. Otherwise there were no statistically significant differences in the rate of adverse event categories across treatment groups. 57 events were rated as probably or definitely related to a pre-existing underlying condition. Of all the adverse events, 125 had resolved and 56 were ongoing.
Adverse events by category and group.
Device-Related Events
Two adverse events were rated as definitely related to the iFuse device. One subject had implant-related impingement on a sacral nerve root requiring immediate revision. Pain resolved promptly on reposition of the device. A second subject developed a hairline (undisplaced) fracture of the ilium adjacent to the caudal-most implant, diagnosed on CT scan, causing buttock pain 3-4 months after the index procedure, possibly related to lifting a heavy object. Pain resolved gradually with conservative treatment. A third subject developed contralateral SI joint pain, which was deemed probably related to the implant as a result of a change in biomechanics related to the placement of the study device (Table 6).
Adverse events related to device, procedure, other treatment for SI joint pain or pre-existing condition.
Procedure-related Events
Seventeen events (16.7% rate) were probably or definitely related to the surgical procedure and 4 events (8.7% rate) were probably or definitely related to SI joint treatments in the NSM group. Events related to the surgical procedure included neuropathic symptoms (n=2, 1 case directly attributed to the iFuse implant itself), postoperative medical problems (n=4, e.g., urinary retention, nausea/vomiting, atrial fibrillation), SI joint pain or trochanteric bursitis (n=4), surgical wound problems (n=5), iliac fracture (n=1) and an asymptomatic physical exam finding (n=1). All events resolved except for 2 subjects with ongoing pain. Three NSM subjects experienced increased back or SI joint pain after physical therapy, SI joint steroid injection or RF ablation (one case each) and another subject had flushing and shortness of breath associated following a SI joint steroid injection. Finally, one subject reported worsening SI joint pain related to postoperative rehabilitation after surgery.
Event Severity
Twenty-seven of the 181 reported events were rated as severe (21 in the surgery group and 6 in the NSM group, p=0.3241). In the surgery group, 2 severe events were device-related (described above) and 4 were procedure-related (one each of wound hematoma, iliac bone fracture, postoperative impingement of the implant on a sacral nerve root and postoperative atrial fibrillation/respiratory failure). The one severe event in the NSM group attributed to NSM was back pain attributed to physical therapy.
Discussion
Back pain is one of the most important causes of disability worldwide56–58 and SI joint pain has been shown to be a common cause of lower back pain.2 Our study enrolled patients with SI joint pain due to degenerative sacroiliitis or sacroiliac joint disruption that was carefully diagnosed using a rigorous definition of disease that combines medical history (including risk factors for SI joint pain), the presence of at least 3 SI joint-directed physical examination findings,10 and confirmation using a diagnostic SI joint block performed within 3 months of enrollment. As evidenced by our subjects’ baseline Oswestry Disability, SF-36 and EQ-5D scores, patients with chronic SI joint pain can be quite debilitated and have a very poor quality of life.50
Because traditional open SI joint arthrodesis is now rarely performed unless direct visualization of the joint surfaces is required,35 this type of procedure was not feasible to use as a control group. Since the optimal non-surgical treatment for chronic SI joint pain is not known, all available non-surgical treatments, especially those for which there is high-quality evidence for at least short-term responses,20–23 were offered in a stepwise fashion to subjects assigned to non-operative care.
Within 1 month after undergoing surgery, subjects assigned to SI joint fusion reported statistically and clinically significant improvements in pain, disability and quality of life. In contrast, subjects assigned to NSM were found to have mean changes that were of minor clinical importance. Clinical success at 6 months occurred in >80% in the surgical cohort as compared to <25% in the NSM cohort (posterior probability of superiority >0.999). Similarly, a larger proportion of surgical subjects experienced a clinically important improvement in disability as measured by Oswestry Disability Index compared to the NSM group (75 vs. 27% with ≥15 point improvement). Changes in quality of life paralleled these findings, with marked improvements in EQ-5D and all domains of SF-36 in the surgical group but small improvements in the NSM group. Pre-planned subgroup analysis showed similar treatment effect sizes across subgroups. The proportion of subjects using opioid analgesics in the surgery group decreased in our present results, while that in the NSM group increased.
The findings from our study are consistent with the results from prior studies. While randomized trials of non-surgical interventions such as SI joint steroid injections20, 21 or RF ablation22, 23 have shown short-term effectiveness, there is no currently published evidence that suggests that these interventions will give rise to long-term relief. Physical therapy, performed according to APTA guidelines and tailored to individual needs, did not appear to be effective during the first 6 months in this patient population. At the time of trial design, a comprehensive review showed no high-quality published literature suggesting that PT is effective in this patient population.59 CBT-based treatments were not used as they were deemed unstandardizable, impracticable and unrepresentative of modern US healthcare. The few published randomized trials of SI joint steroid injections20, 21 used peri-articular injection rather than the intra-articular technique in current use in the US and were limited by very short-term follow-up (maximum 2-month follow-up). Patients in our study were diagnosed with SI joint pain as confirmed by intraarticular anesthetic block. Because of the known multiple innervation of the SI joint6, RF ablation may have been less effective in these subjects. The improvements in pain, disability and quality of life seen in our study's surgically treated subjects at 6 months essentially mirror those observed in multiple other single-center case series,36–42 a combined multicenter analysis,43 and a single-arm prospective multicenter trial44 assessing the iFuse device, including studies that report 2,36, 43 442 and 5-year41 follow-up. Furthermore, the 6-month improvements seen in the surgical group were remarkable given the high prevalence of failure of both prior treatments (72% had received physical therapy and 86% had received SI joint steroid injections) and prior back surgery and competing diagnoses (37% had prior lumbar fusion which is a well-accepted risk factor for degenerative sacroiliitis58 and 15% had a history of spinal stenosis). The presence of concomitant pathology like this can make the diagnosis of SI joint-mediated pain challenging and can limit the overall restoration of health. The high prevalence of prior lumbar fusion also raises the possibility that some patients had previously undergone lumbar spine fusion when a component of their true underlying diagnosis may have included pain arising from the SI joint.
Two mechanisms likely explain the significant pain relief and improvements in disability demonstrated by the surgical group. First, the unique triangular shape of the implants as well as the fact that at least 2 implants were anchored within the cortical surfaces of each side of the joint (i.e. both the ilium and sacrum), would be expected to immediately stabilize the SI joint; stabilization (and subsequent fusion) of joints is a known technique which alleviates pain caused by degenerative arthrosis. Given the altered muscle activation in patients with SI joint pain relative to asymptomatic individuals,9 it is possible that stabilization and fusion of the SI joint may also promote the resolution of altered muscle recruitment and gait abnormalities. Late pain relief likely occurs as a result of biological fixation of bone in both the sacrum and ilium as well as bridging bone across the joint, all of which appear to occur commonly in long-term follow-up.41 Interestingly, neither pain relief nor bridging bone seem to require full decortication of the SI joint as is typically done in other fusion procedures. (CT scans in INSITE subjects, to be performed at 2 years after SI joint fusion, are not yet available and will be reported separately.) The minimally invasive surgical approach used in this study, employing standard orthopaedic principles of joint stabilization followed by long-term fusion, provides the benefits of joint fusion while minimizing surgical morbidity and shortening recovery times.
The iFuse surgical procedure is straightforward. However, as with other instrumentation techniques, such as iliosacral screw fixation,60 the surgeon must pay careful attention to SI joint anatomy and be cognizant of any variations in order to ensure placement of all devices fully across the SI joint. Nevertheless, a clinical case series and a safety review61 suggest that the complication rate of this procedure is relatively low.
Adverse events occurred slightly more commonly (but not statistically significantly) in the surgical compared to non-surgical groups during the first 6 months of follow-up. Device-related adverse events in the surgical group were uncommon, with only one case of impingement of the distal end of the implant on a sacral nerve root and the resultant pain resolved promptly following repositioning of the devices. Procedure-related adverse events mostly involved standard complications that are inherent to all operations. Adverse events in the NSM group were primarily worsening of SI joint pain during physical therapy or related to RF ablation. Most adverse events reported in the study were not related to study procedure or device, and many were attributed to pre-existing conditions. Collection of adverse events will continue through the study's planned 2-year follow-up.
The study had several advantages. For instance, participants were carefully screened using predetermined eligibility criteria and all potential subjects underwent rigorous diagnostic testing including an SI joint block performed within 3 months prior to screening. The proportion of screened subjects who participated was high, suggesting generalizability to the population as a whole. Pain, disability and quality of life questionnaires, which form the basis of most of the analyses in this report, were completed primarily by subjects under the direction of study coordinators, not treating physicians, to minimize any bias theoretically linked to the study's industry sponsorship. In addition, the 6 month follow-up rate was high and no early crossover occurred. This study incorporates the prospective results recorded for multiple surgeons and pain specialists, and the differences in treatment effects were consistent across all measures. The NSM intervention included all modalities for which there is at least some evidence of clinical effectiveness for this condition. The study was executed according to an international clinical trial standard (ISO14155:2011); study data were collected uniformly on electronic case report forms at pre-determined time points and as such were rigorously monitored and source verified.
The study is currently limited by follow-up to six months only; subjects will continue to be followed for 2 years, at which point radiographic outcomes will be able to be reported. While it is possible that NSM could take more than 6 months to reach its full effect on pain, disability and quality of life, this seems unlikely, as response rates to date are low and many participants had already undergone one or more of the treatments used in the NSM prior to enrollment. Currently, more than three quarters of NSM subjects crossed over to surgical SI joint treatment, limiting the ability to derive unbiased across-treatment comparisons after the 6-month visit. At the time of trial design, investigators believed that a post-market surgery vs. non-surgery trial of this technology in the US would have been very challenging to enroll in a reasonable time frame without a crossover component for subjects who did not derive relief from NSM. In other studies, long-term follow-up (at two36, 43, four42 and five41 years) of patients undergoing SI joint fusion with the same device has shown sustained positive outcomes.
Combined with prior published evidence, this study confirms that minimally invasive fusion of the SI joint using triangular implants provides superior six month outcomes compared to non-surgical management in patients with chronic SI joint dysfunction due to degenerative sacroiliitis and SI joint disruption and may therefore be a preferred treatment option for patients who have failed to respond adequately to conservative therapies. In this study, NSM delivered over course of 6 months provided only minor relief of pain and disability in patients with chronic SI joint pain unresponsive to conservative treatment.
Conclusions
In patients with severe SI joint dysfunction due to degeneration or disruption of the joint, minimally invasive SI joint fusion using triangular implants placed across the joint provides superior six month outcomes compared to non-surgical management.
Disclosures
All authors conduct clinical research for SI-BONE. The study was sponsored by SI-BONE (San Jose, CA). Peter Whang is a paid SI-BONE consultant participating primarily in educational events. Clay Frank is an SI-BONE consultant participating primarily in educational events, but receives only reasonable expense reimbursement as compensation. Harry Lockstadt has a consulting agreement with SI-BONE, but has not provided or been paid for services as of the date of this article. Daniel Cher is an SI-BONE employee.
Acknowledgements
The authors acknowledge the 59 investigators and coordinators in the INSITE Study Group: John T Cummings Jr. MD, Elizabeth Pertile (Community Neurosurgery, Indianapolis, IN); Clay Frank MD, Tracy Mente RN (Wheaton Franciscan Healthcare, Wauwatosa, WI); Scott Kitchel MD, Shawn Potts (Neurospine Institute, Eugene, OR); Robert Limoni MD, Nilesh Patel MD, Taylor McGinnis, Nicholas Peterson (Aurora BayCare Medical Center & Advanced Pain Management, Green Bay, WI); Harry Lockstadt MD, Elaine Wilhite MS, James Farris PA-C (Bluegrass Orthopaedics & Hand Care, Lexington, KY); Don Kovalsky MD, Laura Pestka RN (Orthopaedic Center of Southern Illinois, Mount Vernon, IL); Peter Whang MD, Donna Ann Thomas MD, Bethany Samperi, Stacey Lombardi (Yale University, New Haven, CT); John A. Glaser MD, Emily A. Darr MD, Laura Fields, Jennifer Philp (Medical University of South Carolina, Charleston, SC); Charles Harvey, MD, Jason Peterman PA-C, Karim Bouferrache MPAS PA-C, Lori Latham (Riverside Medical Center, Kankakee, IL); Pierce Nunley MD, Andrew Utter MD, Marcus Stone PhD, Norma Rivera (Spine Institute of Louisiana, Shreveport, LA); Ed Santos MD, David Polly MD, Jonathan Sembrano MD, Charles Ledonio MD, Sharon Yson MD (University of Minnesota, Minneapolis, MN); Philip Ploska MD, Terry Price PA (Resurgens Orthopaedics, McDonough, GA); Michael Oh MD, Gary Schmidt MD, Matthew Yeager (Allegheny General Hospital, Pittsburgh, PA); Merle Stringer MD, Douglas Stringer MD, Carolyn Henderson (Brain & Spine Center, Panama City, FL); Farshad Ahadian MD, Yu-Po Lee MD, Katie Lam (University of California, San Diego, CA); Gowriharan Thaiyananthan MD, Bryan Oh MD, Navid Farahmand MD, Tungie Williams (Basic Spine, Newport Beach, CA); William Rosenberg MD, Laura Textor, APRN (Midwest Division-RMC, LLC,-Research Medical Center, Kansas City, MO); Vikas Patel MD, Scott Laker MD, Venu Akuthota MD, Claire Cofer (University of Colorado, Aurora, CO); Mark C. Gillespy MD, Sherri Zicker RN (Orthopaedic Clinic of Daytona Beach, Daytona Beach, FL).
The authors acknowledge the SI-BONE clinical affairs team (Kathryn Wine, Corinne Lee, Denise Law, Jeff Price, Shira Stone, Terrill Himmelmann) for help with study conduct and monitoring.
- Copyright © 2015 ISASS - International Society for the Advancement of Spine Surgery
Copyright © 2015 ISASS.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Minimally invasive sacroiliac joint fusion using triangular titanium implants versus nonsurgical management for sacroiliac joint dysfunction: a systematic review and meta-analysis
- Minimally Invasive Sacroiliac Joint Fusion vs Conservative Management in Patients With Sacroiliac Joint Dysfunction: A Systematic Review and Meta-Analysis