


Abstract
Standards are important tools in evaluating and predicting the performance of medical devices prior to implantation. There are three types of standards that are available: a material specification, a standard test method, and a standard test guide. Each of these types of standards is defined with examples of how each is used to facilitate evaluation of medical devices. The standards development process is also described: this is a complex process, requiring the involvement of a multidisciplinary team, usually consisting of engineers, scientists, and clinicians who represent healthcare, academia, government, and industry. Finally, standards have a clear and defined role in the development of medical devices, and the benefits, strengths, as well as the limitations in this role are discussed.
Introduction
Have you ever wondered how a device company is able to compare a device to a similar device marketed by a competitor? Why can one manufacturer claim that a particular device is “stronger” or “stiffer” than that other device? In many cases, the manufacturer makes claims about the product in relative terms compared to other products, and many products are cleared or approved by FDA by making useful comparisons to existing products. Typically, the manufacturer is using some mechanical testing data to justify any comparative statements and claims about a particular device. The process by which these data are generated, however, is often difficult for users of the device to understand, particularly the process by which the manufacturers determined which tests to conduct to obtain the data. If each manufacturer tested their device differently, there would be no way to compare devices to make objective evaluations on their performance characteristics. Logically, some common ground is needed so that devices can be objectively evaluated. To this end, standards are needed, and each of us in the medical device field is accustomed to manufacturers or users of spinal implant devices asserting that a device “conforms to a standard” or the device was tested “according to a standard.” The purpose of this article is to discuss the need for standards, the development process, and their strengths and limitations.
Different types of standards
There are 3 main types of standards that are relevant to spinal implants and other medical devices: standard material specifications, standard test methods, and standard test guides. These types of standards serve very different purposes.
Material specifications actually list chemical and physical properties that a material must have in order for a material supplier to claim that the material meets the standard. ASTM International (formerly the American Society for Testing and Materials), for example, publishes many standard specifications for different metallic alloys such as titanium, stainless steel, or cobalt chrome for surgical applications. ASTM F136-08, a standard specification for titanium alloy, describes the maximum percent compositions of each chemical element (eg, 0.05% nitrogen, 0.08% carbon), as well as minimum strength values that the material must meet in order to conform to the standard. Standards like these are published for most of the major surgical alloys and metals, as well as for medical polymers such as ultrahigh molecular weight polyethylene (UHMWPE), acrylic bone cement, and polyetheretherketone (PEEK).
Standard test methods describe how to set up and conduct a test of a specific mechanical property or device characteristic. For a mechanical test, for example, the standard might describe how to grip the device in the testing machine, where and how loads should be applied, and how to build any test blocks or fixtures that are required. In many cases, a standard test method will actually include multiple sub-methods for testing in different loading modes such as compression, shear, or torsion. Specific test parameters, such as the number of samples to be tested or the amount of load, are specified, as well as a description of the measurements or properties that should be reported as results. However, standard test methods for spinal devices do not generally define any specific values that the results must meet in order for a device to “pass” the test. These types of standards are commonly referred to as performance standards, as they define performance levels for a given device. Most standards do not define performance criteria, but rather leave this responsibility to the user to define his or her own acceptance criteria for each test based on the intended application of the device. Although standard test methods do not usually define performance criteria for a device, the test method itself is beneficial, because it defines a protocol for producing repeatable, reliable results and avoids the need for every manufacturer to invent a new test method each time a new product is being developed. In this way, standard methods allow comparisons of data not only among different device designs tested in the same lab but also devices tested at different labs.
Finally, a standard test guide usually precedes the development of a test method. Typically, when the process of developing a new standard is beginning, the devices that are intended to be tested are still undergoing early clinical evaluation, and their in vivo performance and potential failure modes are unknown. In cases such as these, the standard will be written in a more general fashion specifying tests and methods that should be evaluated, but stopping short of delineating precise test methods. As more experience is obtained through “bench-top” testing and clinical evaluation, a formal standard test method can be written. This is usually a joint effort between clinicians and engineers who collaboratively tailor the standard test method to correspond to how the implants perform in vivo.
It is important to understand, therefore, that if a spinal implant device's material “meets a standard,” that statement tells you something very specific about the material's chemical and physical properties. If a spinal implant device was “tested according to a standard,” then this statement means that an established test method or guide was used in order to evaluate the device, but more information is needed if you want to know how the results of that testing compared to some objective criteria.
How standards come to be (Who writes standards?)
Standards development is both a voluntary and consensus process. In addition to ASTM International's F04 Committee on Medical and Surgical Devices, other organizations, including the International Standards Organization (ISO) and Association for the Advancement of Medical Instrumentation (AAMI), also have groups devoted to medical device standards. The development of a standard is a consensus process with people working together to agree on a standard method or specification. This is a complex endeavor and often requires teamwork between engineers, scientists, and clinicians. Because of this multi-disciplinary task at hand, these groups are made up of volunteer representatives from healthcare, academia, government, and industry who work together to write a standard method or practice. It should be noted that the activity of designing appropriate tests and standards for device characterization requires highly skilled individuals who have the ability to bridge the gap between clinical and scientific perspectives. An extensive understanding of anatomy, pathology, biomechanics, and engineering, which is usually gained through cross-functional teams, is critical to successful standards development.
Standards organizations have voluntary membership and have no legal authority to impose or enforce the implementation of their standards. Some standards have been adopted by some governments as part of their legislative or regulatory framework, but such decisions are made by individual governments and not by the standards organizations. In the United States, the Food and Drug Administration (FDA) has a standards recognition program by which consensus standards may be evaluated by the FDA and recognized for use in satisfying a regulatory requirement. This conformity is voluntary but intended to reduce the time and burden necessary for clearance or approval of a device, as both the manufacturer and FDA are already familiar with the details of the standard.
Benefits of standards (Why do we use standards?)
The primary benefit of a successful standard test method is that it establishes a procedure that can be used by different laboratories to test different devices and obtain results that can be meaningfully compared. In theory, standards should also establish consensus in the scientific community as to the best currently available test procedures for a specific type of device. A standard should ideally be supported by experience and data obtained from testing so that users have some confidence that the techniques described can produce repeatable, reliable results. As discussed previously, if a standard test method has been developed for a particular type of device, manufacturers should be spared from having to “reinvent the wheel” each time they develop a new design. To put it a different way, if 12 companies are developing the same type of device, it should be more efficient if they can each use an already-published standard test method, instead of having to spend extra time in the lab designing 12 different test protocols from scratch.
Another important benefit of a standard test method is that it streamlines the amount of information that must be communicated between a manufacturer who performs the testing and someone else who is interested in the results. If the 2 parties are both familiar with the published standard method, then the manufacturer does not need to explain the technical details of the testing but can instead focus on presenting and discussing the results. Because the test setup and testing parameters should already be familiar to both parties, everyone begins with a common point of reference.
Examples of current spine standards
Table 1 lists some of the material specifications and standard test methods that relate to spinal implant devices, as currently published by ASTM International and ISO. These 2 organizations have published standards that address most of the major categories of spinal device: fusion systems, interbody fusion devices, disc replacements, and other nonfusion devices or systems. For fusion systems, these standards describe testing of mechanical strength, stiffness, and fatigue strength of individual components or entire systems. For interbody fusion devices, also known as cages, these standards describe static and dynamic testing of cages as well as measuring the resistance of a cage to subsidence (i.e., the device sinking into the vertebral body endplate). Standards have also been created to address static, fatigue, and wear testing for total disc replacements. ASTM has also published 2 standards describing testing of other types of nonfusion systems, specifically total facet replacements and extra-discal motion preserving systems. Because of the recent rapidly-growing interest in motion preservation and dynamic nonfusion devices, there are many types of devices for which standards are still being developed but for which none currently exist. For those devices, a manufacturer may be able to begin with a published standard for a similar device and adapt it as necessary for their design, or more work may be needed to develop a battery of original test methods that addresses all potential failure mechanisms.
Examples of standard material specifications and test methods relevant to spinal implant devices
The process of standards development
Standards development usually begins with the product development process, since new products need to be tested. Typically, surgeons identify persistent clinical problems and these unmet needs spawn the innovation of new, novel techniques and implants to treat spinal disorders. But will these new devices work clinically? Part of the process for determining how a new implant will perform is to perform a design failure modes and effects analysis (DFMEA) to evaluate relevant modes of failure of the device (i.e., how might it fail in vivo in its intended application?). This analysis, complemented by an understanding of the in vivo stresses and strains to which the implant will be subjected, allows the engineer to develop relevant test protocols that evaluate the potential failure modes of the device. In most cases, this process requires multiple tests, test configurations, and test fixtures. If a consensus can be reached, these individual test methods can eventually become standards.
To cite an example of the development of a standard, a recent test method that was published by ASTM subcommittee F04.25 relates to the testing of extra-discal motion preserving devices (F2624-07, Standard Test Method for the Static, Dynamic, and Wear Assessment of Lumbar Extra- Discal Spinal Motion Preserving Implants). These systems can take several forms, including pedicle screw-based systems with cords or flexible rods or devices that act as spinous process “bumpers” which limit spinal extension. Because no applicable standards existed when these products were in the early stages of development, each engineer had to modify existing standards or develop their own test method to evaluate their own device. The engineers who had already been testing these types of devices assisted the standards process as their data and experience were available to facilitate the drafting of a standard test method. The subcommittee agreed early in the development of the standard that it would provide test methods for the static, dynamic, and wear testing of extra-discal motion preserving implants.
While agreeing on the scope was relatively easy, coming to a consensus of how to actually perform these tests required a collaborative effort between engineers and surgeons. After 2 and a half years of discussions and testing, the ASTM subcommittee reached a consensus on how the tests should be performed. Figure 1 shows the evolution of the fixtures for testing extra-discal motion preserving devices during the development of the test method. Initially, a stainless steel ball and a socket machined into 2 simulated vertebral bodies were proposed (Fig. 1A) to mimic the rotations of the spine. While this was a simple apparatus, it was agreed that isolating wear debris from the device (as opposed to debris generated from the ball and socket) would be too difficult. The next iteration of the fixture design was overly complex, employing a series of rocker arms to generate flexion/extension motion (Fig. 1B). The final version (Fig. 1C) relies on a torsional actuator to generate flexionextension motion, effectively allowing the engineer to control the moments to which the device is subjected while allowing for particle isolation.
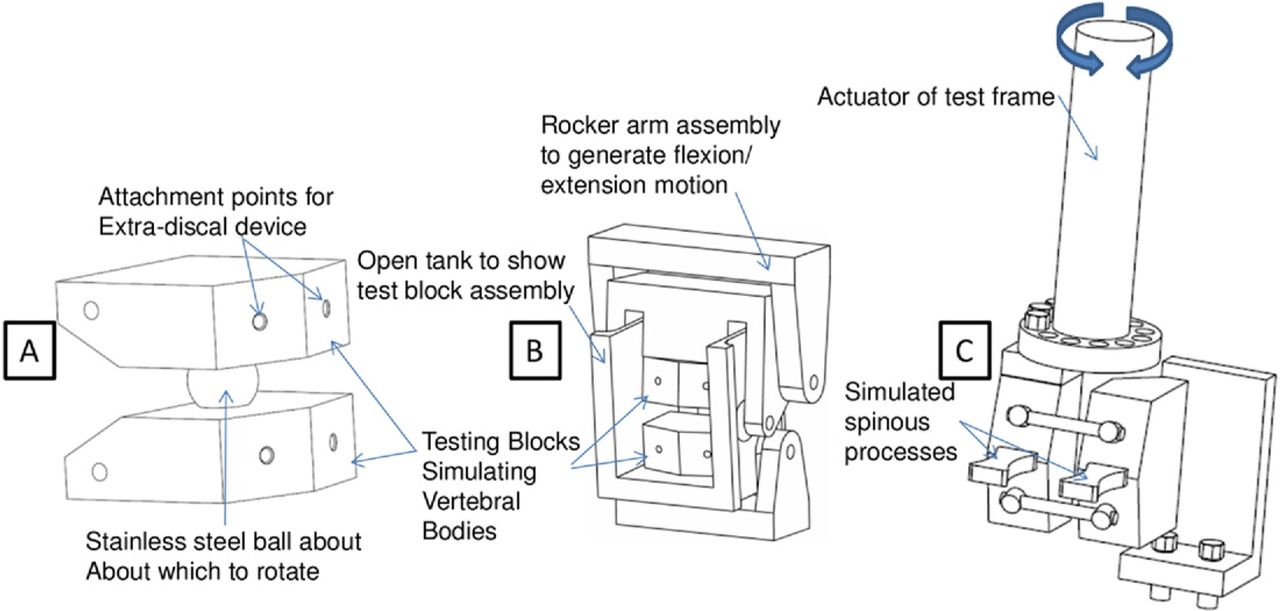
The evolution of flexion/extension test configurations (A) initial concept using a stainless steel ball about which to rotate to mimic the rotations of the lumbar spine, (B) intermediate concept employing external rocker arms to generate flexion/extension motion, and (C) final concept drawing of assembly used in extra-discal standard.
The development of F2624 serves as a good example of the iterative process by which consensus is reached on a standard. The use of this standard provides a common denominator and effectively facilitates device comparison based on static, fatigue, and wear characteristics in flexion/ extension, lateral bending, and axial rotation. This test method provides data to the investigator, which can then be used to decide whether a particular device could be used clinically, or, in many situations, as a starting point for the next design iteration of the product. It is also important to understand that standards are intended to be “living” documents that are updated as new information becomes available. This updating occurs by 2 pathways: (1) at any time, a member can identify or generate new data that should change a standard and present a draft of new language for balloting; or, (2) alternatively, changes can be made during a periodic review and reconfirmation process which occurs every 3–5 years depending on the standards organization. ASTM standards, for example, are required to be reviewed and reapproved every 5 years. While F2624 serves as a good example of the iterative process by which standards are developed, the final verdict on the usefulness and applicability of this particular standard relative to the in vivo performance of extra-discal motion preserving devices is yet to be determined. In some instances, standards must be significantly revised to provide meaningful data that is indicative of successful in vivo performance. One way that standards organizations facilitate review of standards is by sponsoring symposia and workshops to call for papers regarding current standards, so that they may be evaluated for effectiveness and updated as appropriate, or, in some cases, deleted altogether. Regardless of the method for how standards are updated, it is crucial to the success of any standards organization for standards to remain up to date so that standards remain meaningful and useful to the scientific and medical community.
Limitations of standards (What can't they do?)
As previously discussed, a limitation of standard methods is that they do not dictate how to interpret testing results or whether a particular result should be considered a “success” or “failure.” It is the responsibility of the user to define the acceptance criteria for the test and to compare the final results to these acceptance criteria to determine whether or not the device should be suitable for the desired application. Acceptance criteria for mechanical testing of spinal implants are generally developed from 2 kinds of sources. Because the FDA's 510(k) notification process requires some devices to be shown “substantially equivalent” to a previously-cleared device, acceptance criteria may often be based on data from another device. An alternative approach is to use acceptance criteria based on the expected physiologic loads or motions that will be applied to the device in vivo, based on the various estimates of spinal loads and kinematics that can be found in the biomechanics literature. However, interpretation of the biomechanical literature can be very subjective, and requires that literature data are applicable to the particular location, application, and loading mode of the device. As a result, establishing robust acceptance criteria based on biomechanics may be significantly more complex than using a comparable device.
A standard test method may not always work as a “one size fits all” method, and may require slight modification or adaptation by a user to fit his or her particular new design. A standard must be specific enough to evaluate a type of device with good reproducibility, yet it must be defined broadly enough to allow testing of more than just one particular design. Innovation and competition demand that no 2 devices be exactly the same in terms of geometry, materials, and other design characteristics. Therefore, standards must be flexible enough to evaluate and compare different designs, yet not so open-ended that they prohibit meaningful comparisons among them. If a standard must be modified, the user must realize that some modifications will have greater consequences than others, and understand how each modification of the method will affect her ability to compare the results with data from devices tested using the original, unmodified method. In reporting results of a modified standard method, it is also important that the user report what modifications were made and the justification for each, so that others may understand his rationale.
Finally, and most importantly, it should be recognized that standards are typically focused on methods of measuring very specific characteristics of a device such as strength, stiffness, or wear resistance, and are not intended to duplicate all of the complex, multi-axial, weight activity-dependent loads on the spinal column. It is difficult to simulate the complex in vivo loading environment in an in vitro test, but clinical experience with spinal implants and biomechanical research can guide the development of simpler tests focused on specific mechanical properties. These properties can be used to compare different devices to each other and to anticipated loads. Test results provide useful tools for comparison of devices, but results cannot necessarily be extrapolated to predict clinical performance because of the complexity of the in vivo environment compared to rigidly controlled laboratory conditions. In the previous example of ASTM F2624, a standard was developed that effectively compares mechanical differences between devices, but whether those differences translate into differences in clinical performance remains to be seen. Typically, that correlation between mechanical and clinical differences can only be investigated once there have been enough devices explanted to examine their failure modes and compare them to devices tested according to the standard. Again, while F2624 was reached by a consensus effort of an ASTM sub-committee, ultimately, data from retrievals will serve to validate whether the methods detailed in F2624 are appropriate for evaluating extra-discal motion preserving devices. In this same light, some analyses of retrieved total disc replacements, for example, have suggested modifications that should be made to future versions of the existing disc wear standards.1–3
Clinical failure modes, challenges, and conclusions
It is incumbent upon an engineer developing a product to address all potential failure modes. Testing a device using one or more standard test methods is prudent. However, in light of all of the potential in vivo failure modes, using only standard test methods to evaluate a potential product would be a mistake. There are indeed in vivo failure modes that cannot be addressed through bench-top testing, particularly those that are related to biological responses that cannot be mimicked in an in vitro environment. Examples of in vivo failure modes that would be difficult to predict using benchtop models are device expulsion, device-related osteopenia, and subsidence. Because these potential failure modes are a function of the complex biological and biomechanical environment of the implants, which the investigator cannot duplicate precisely in the lab, assessment of these failure modes requires other modes of evaluation.
To build on the previously-discussed example of the extra-discal motion preserving device test method, wear debris from the test bath could be characterized using the standard, but making definitive statements regarding the biological effect of these particles would be difficult without further study. Might the particles result in an immune response, ultimately resulting in a loose implant unable to stabilize the spine, or would the particles result in chronic inflammation and pain even if the device stabilized as intended? If particles are suspected to be generated, then separate biocompatibility standards, such as ASTM F1903- 98, F1904-98, F1905-98 or F1906-98, should be applied. Only these types of assessments using animal models would start to answer questions of biocompatibility. Aside from biological issues, fixtures employed for bench-top testing do not recreate the complex loading of the spine, especially since engineers will often simplify the loading to facilitate testing and comparisons between devices (see Fig. 1). The results of bench-top testing based on engineering fundamentals and the expected in vivo biomechanical environment must, therefore, be extrapolated upon to predict in vivo performance. Because assumptions will always play a role in predicting performance, standards cannot, nor should they, ultimately be solely relied upon to determine whether or not the device should be used clinically.
As surgeons, engineers, and scientists better understand the biological and mechanical environment in which implants are intended to function, standards will continually evolve to better address the needs of the medical device community. Other tools, such as mathematical models, that incorporate both the biological response and the biomechanical environment can be employed, and perhaps even developed into standards, hopefully leading to quicker and more successful product development. Other evaluation methods are also required to render a full understanding of how a device performs. These may include in vivo animal experimentation, material, custom-mechanical, biomechanical, histological, clinical, and explant analyses. Regardless of tools used and methods employed, the investigator should always keep in mind that the goal of testing is to evaluate the device's ability to withstand physiologic conditions and function as intended without failure. Clearly, standard test methods are only 1 tool to help achieve this goal and are, consequently, only 1 piece of the puzzle in device characterization. Hopefully, the use of standards will improve communication between those who test devices and also work in concert with other analysis tools to assist the investigators in a full performance characterization of the device.
Footnotes
The ASTM and ISO standards mentioned in this article are published on an annual basis and are available for purchase (in print or electronic editions) via the organizations’ websites, www.astm.org and www.iso.org.
- © 2009 SAS - The International Society for the Advancement of Spine Surgery. Published by Elsevier Inc. All rights reserved.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Different types of standards
- How standards come to be (Who writes standards?)
- Benefits of standards (Why do we use standards?)
- Examples of current spine standards
- The process of standards development
- Limitations of standards (What can't they do?)
- Clinical failure modes, challenges, and conclusions
- Footnotes
- References
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.