


Abstract
Background Elastomeric disc replacements have been developed to restore normal shock absorption and physiologic centers of rotation to the degenerated disc. The Physio-L Artificial Lumbar Disc is an elastomeric disc which uses a compliant polycarbonate-polyurethane core with enhanced endurance properties. The objective of this study was to evaluate the safety and efficacy of the Physio-L through a 12-month follow-up period in a prospective, nonrandomized clinical trial.
Methods Twelve patients who met the inclusion/exclusion criteria were enrolled in the study. Eight patients received a single implant (L5-S1) and 4 received a 2-level implantation (L4-5 and L5-S1). Patients were assessed preoperatively and postoperatively at 6 weeks and 3, 6, and 12 months. Primary outcomes included the VAS, ODI, a radiographic analysis of implant condition, incidence of major complications, and reoperations. Secondary outcomes included SF-36, ROM at index and adjacent levels and disc height.
Results All patients completed the 12-month follow-up evaluations. Through 12 months, the Physio-L devices have remained intact with no evidence of subsidence, migration, or expulsion. VAS low-back pain and ODI scores improved significantly at all follow-up periods compared to preoperative scores. The range of motion of 13.3° ± 5.5° at the index level was considered normal. Overall, patients were satisfied with an average score of 83.5 ± 26.8 mm. When comparing the device to other artificial discs, the current device showed a clinically relevant improvement in both ODI and VAS scores at all follow-up time points. Statistically significant improvements in both scores were observed at 12 months (P < .05).
Conclusion The Physio-L is safe and efficacious, as demonstrated by improved pain relief and functional recovery without any implant failures, significant device related complications, or adverse incidents. The clinical results for VAS and ODI were superior to other marketed artificial lumbar discs such as the Charité and ProDisc-L at the same follow-up timeframes.
Artificial disc replacements (ADR) have been under development for over 20 years for use as motion preservation alternatives to fusion in the treatment of chronic disabling low back pain caused by degenerative disc disease (DDD). Current designs of spinal disc prostheses typically achieve their desired motion by having one surface slide relative to another in a similar manner to total hip and total knee prostheses.1–6 These rigid sliding surfaces are constructed from metal, polymer, or ceramic with differing types of motion dependent on specific designs. It is clear, however, that many of these designs lack resistance to motion and the ability to provide shock absorption. In recent years, mounting concern has been registered in the literature concerning accelerated facet joint degeneration at the index level, an increased rate of adjacent level degeneration after ADR, and stress fractures of the pars or pedicle at the index levels. These untoward effects may be related to the nonphysiologic nature of the design of these disc prostheses.7–10
To overcome these concerns, the use of elastomeric disc prostheses has been proposed to mimic physiologic levels of shock absorption and flexural stiffness; however, an earlier elastomeric disc design using a polyolefin core was developed and examined in clinical trials but was eventually abandoned due to material failure.11, 12 With the recent availability of better fatigue-resistant and bio-stable polymers, a new generation of elastomeric disc prostheses has been developed. The current study evaluates the safety and efficacy of a new generation disc replacement through the 12-month follow-up period.
Materials and methods
The disc prosthesis
The Physio-L (Nexgen Spine, Whippany, NJ) lumbar artificial disc uses a compliant polycarbonate polyurethane that is securely attached to 2 titanium endplates by injection molding the polymer through perforated plates. This provides a purely mechanical attachment that employs no adhesives. This design allows the restoration of the normal range of motion and function of a healthy disc to the involved level (Fig. 1). The domed endplates are manufactured from medical grade titanium alloy and are porous coated with titanium beads to promote bone in-growth and long-term prosthesis-bone interface stability.

Physio-L artificial lumbar disc.
Study design
A prospective, nonrandomized clinical trial was conducted on 12 patients at 2 clinical sites to evaluate the safety of the artificial lumbar disc. All surgeries were performed by 1 of 2 surgeons between March and August 2007. Patients presenting with low-back pain caused by degenerative disc disease (DDD) were enrolled in the study after failing to respond to nonoperative treatment for a minimum of 6 months. Degenerative disc disease at 1 or 2 levels between L3-S1 was confirmed by patient self-reporting of back pain, MRI, and discography. All patients reported in this group fit within defined inclusion/exclusion criteria (Table 1). Contraindications included active systemic infection or localized infection near the implant site, isolated radicular compression symptoms due to disc herniation, allergy or sensitivity to implant materials, osteoporosis, lumbar stenosis, facet joints arthritis, osteopenia, pars defects, instability and/or deformity. Patient evaluations occurred preoperatively (within 3 months of surgery) and postoperatively at 6 weeks and 3, 6, and 12 months. Patients will be followed subsequently at 24 months.
Inclusion/exclusion criteria
Patient demographics
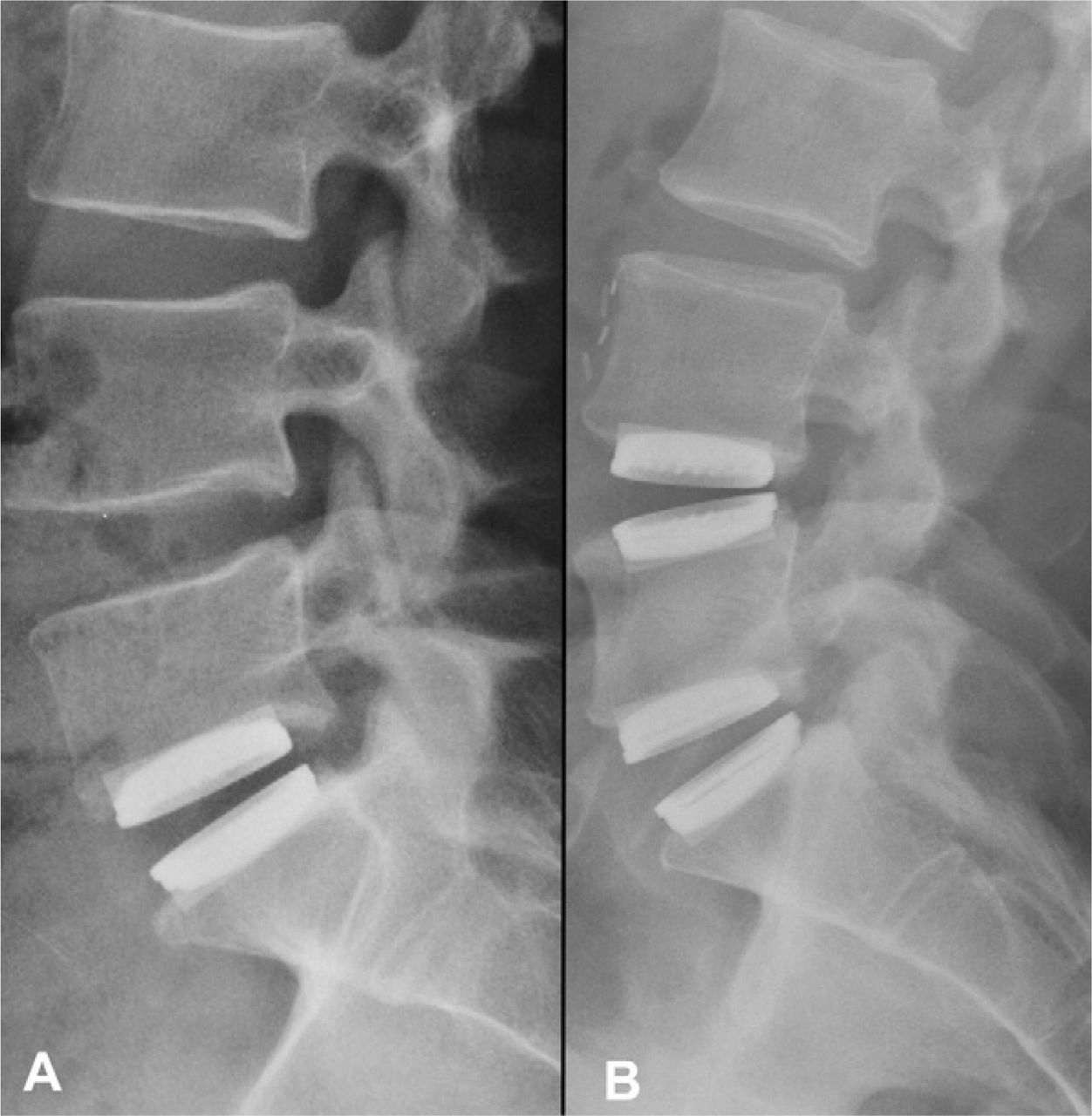
Eleven patients were male and 1 was female, with an average age of 40.6 ± 8.4 years (range, 25–55) and an average BMI of 26.3 ± 3.5 (range, 20.9–31.6). Of these patients, 8 received a single implant (L5-S1 level) and 4 received a 2-level implantation (L4-5 and L5-S1) (Fig. 2). A total of 16 artificial discs were implanted. All patients returned for each follow-up visit up to 12 months.

Lateral radiographs showing A) a patient implanted with a single Physio-L at L5-S1, and B) a patient implanted with two Physio-L devices at L4-L5 and L5-S1 at the 12 month follow up time point.
Surgical technique
The patient was positioned in the supine position and underwent anterior disc removal through an anterior retroperitoneal exposure of the lumbo-sacral spine, which is similar to other artificial disc replacement surgeries. Endplate preparation was performed using contoured bone rasps to closely match the specific dome shape of the metal endplates. A keel cutter was used to cut the channels on vertebral endplates for the central keel without violating the anterior cortex. Following endplate preparation, the artificial disc was inserted as a single unit.
Clinical outcome measures
Patient self-assessment outcome measures included the oswestry disability index (ODI), the visual analog scale (VAS) for back pain, and the Short Form 36 (SF-36) Health Survey questionnaire. A 10-point decrease in ODI scores and 18-point decrease in VAS scores were considered a minimal, clinically important difference (MCID).13 Mental and physical component summary scores (MCS and PCS, respectively) were calculated from the SF-36 Quality of Life questionnaires. A 5-point increase in SF-36 scores was considered a clinically significant improvement.14 Additionally, work status was collected at all follow-up evaluations and patient satisfaction was collected at 6 months and 12 months.
Radiographic analysis
Radiographs were analyzed using an independent radiologist and QMA™ Software (Medical Metrics, Inc., Houston, TX) to evaluate flexion/extension range of motion, disc height, loosening, subsidence, migration, and expulsion.15
Single versus 2-level implantation
Eight patients received the artificial disc at a single L5-S1 level and 4 patients received a 2-level L4-S1 implantation. Patient self-assessment scores of VAS back pain and ODI were compared between these 2 groups.
Comparison to other devices
The present study was conducted using similar protocols and outcome measures to previous reports involving the Charite (DePuy Spine, Raynham, MA) and ProDisc-L (Synthes, West Chester, PA) devices.16–19 As a result, direct comparison of the clinical results for the Physio-L to these devices and their fusion controls is possible. Because no individual subject data was available for the referenced studies, only the reported means and standard deviations of the ODI and VAS scores were used for the comparisons, using 1-way ANOVA (applied to summary statistics). The fusion groups from the Charite and Prodisc-L studies were pooled in the analysis.
Results
Surgery
Mean operative time was 157.5 ± 46.3 minutes (range, 90–240), mean total blood loss was 439 ± 555 cc (range, 70–2000), and the mean average length of hospital stay was 1.2 ± 0.4 days (range, 1–2) for all patients regardless of the number of levels treated. Surgical data was separated to determine the effect of patients treated for single level or 2-level disc disease and results are reported in Table 2.
Operative data for single and two-level Physio-L implantations
Using a paired t test, no significant differences were observed in the total operative time, blood loss, or length of hospital stay for patients receiving a single level replacement versus a 2-level replacement (P < .05). Two approach- related adverse events were recorded during or immediately after the surgery. One patient receiving a 2-level implantation experienced clinically significant blood loss greater than 1500cc, requiring transfusion. This adverse event was resolved without further incident. One patient experienced retrograde ejaculation between the 3- and 6-month follow-up evaluations, and this event was resolved spontaneously by the 12-month follow-up evaluation.
Clinical outcomes
ODI
Mean preoperative ODI scores were 54.3 ± 11.1, and decreased to 17.2 ± 14.6, 15.8 ± 13.9, 13.5 ± 13.9, and 12.7 ± 14.8 at 6 weeks and 3, 6, and 12, respectively. This corresponds to an overall decrease of 75.2% at 6 months and 76.7% at 12-month follow-up (Fig. 3). A statistically significant decrease in mean ODI scores (P < .05) was calculated at all follow-up evaluations when compared with the preoperative scores. Additionally, these results indicate that a clinically relevant difference in function has occurred when comparing the preoperative to all postoperative scores, with a mean decrease in ODI greater than 10 points.
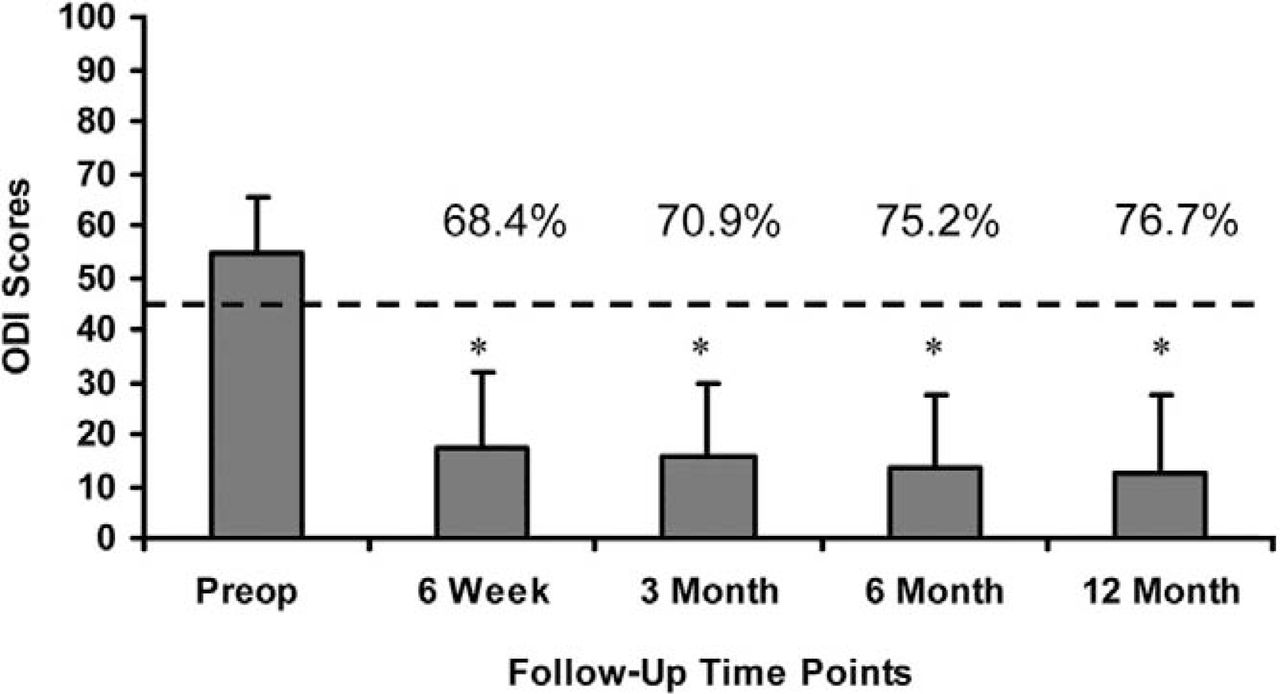
Mean ODI scores at all follow up evaluations. Error bars indicate standard deviations. * indicates statistically significant differences (p < 0.05) from the pre-operative scores. The dotted line indicates the minimal clinically important difference (MCID) of a 10 point decrease from the pre-operative score.
VAS
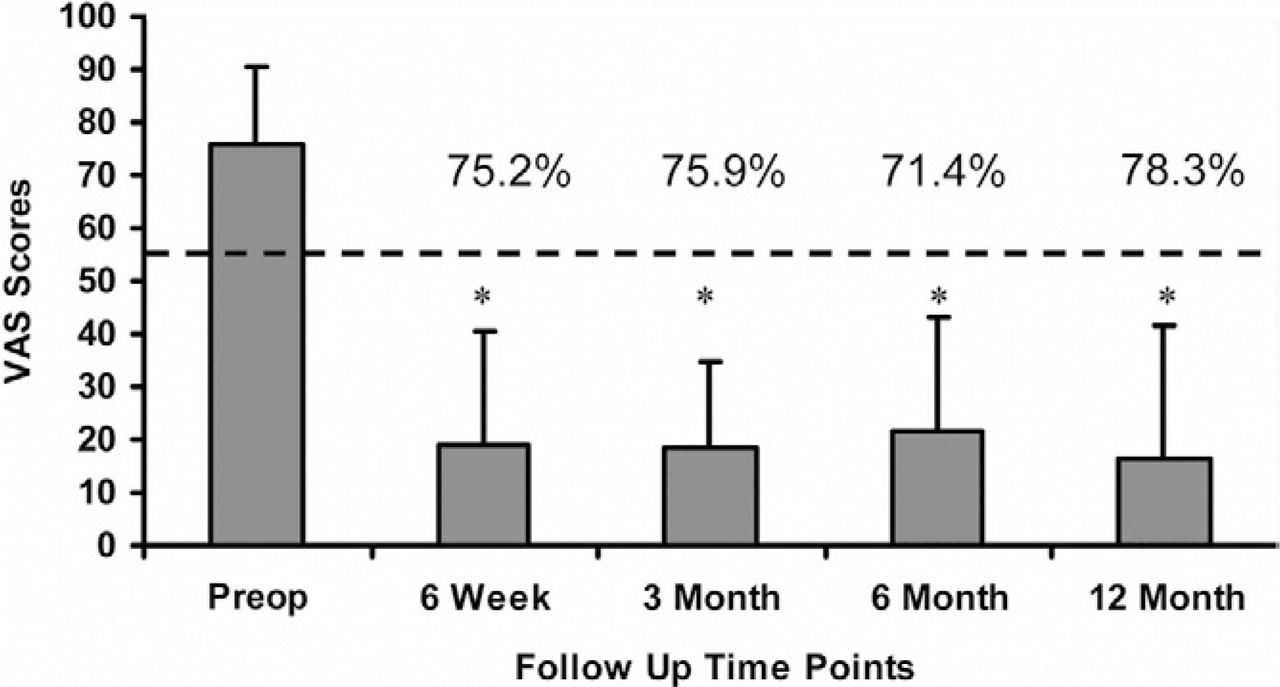
Mean preoperative VAS scores were 76.0 ± 14.3 and decreased to 18.8 ± 21.4, 18.3 ± 16.3, 21.8 ± 21.5, and 16.5 ± 25.1 at 6 weeks and 3, 6, and 12 months, respectively. This corresponds to an overall decrease of 71.4% at 6 months and 78.3% at 12-month follow-up (Fig. 4). A statistically significant decrease in mean VAS scores (P < .05) was calculated at all follow-up evaluations when compared with the preoperative scores. Additionally, these results indicate that a clinically relevant difference in low back pain has occurred when comparing the preoperative to all postoperative scores with a mean decrease in VAS greater than 18 points.

VAS scores at all follow up evaluations. Error bars indicate standard deviations. * indicates statistically significant differences (p < 0.05) from the pre-operative scores. The dotted line indicates the MCID of an 18 point decrease from the pre-operative score.
SF-36
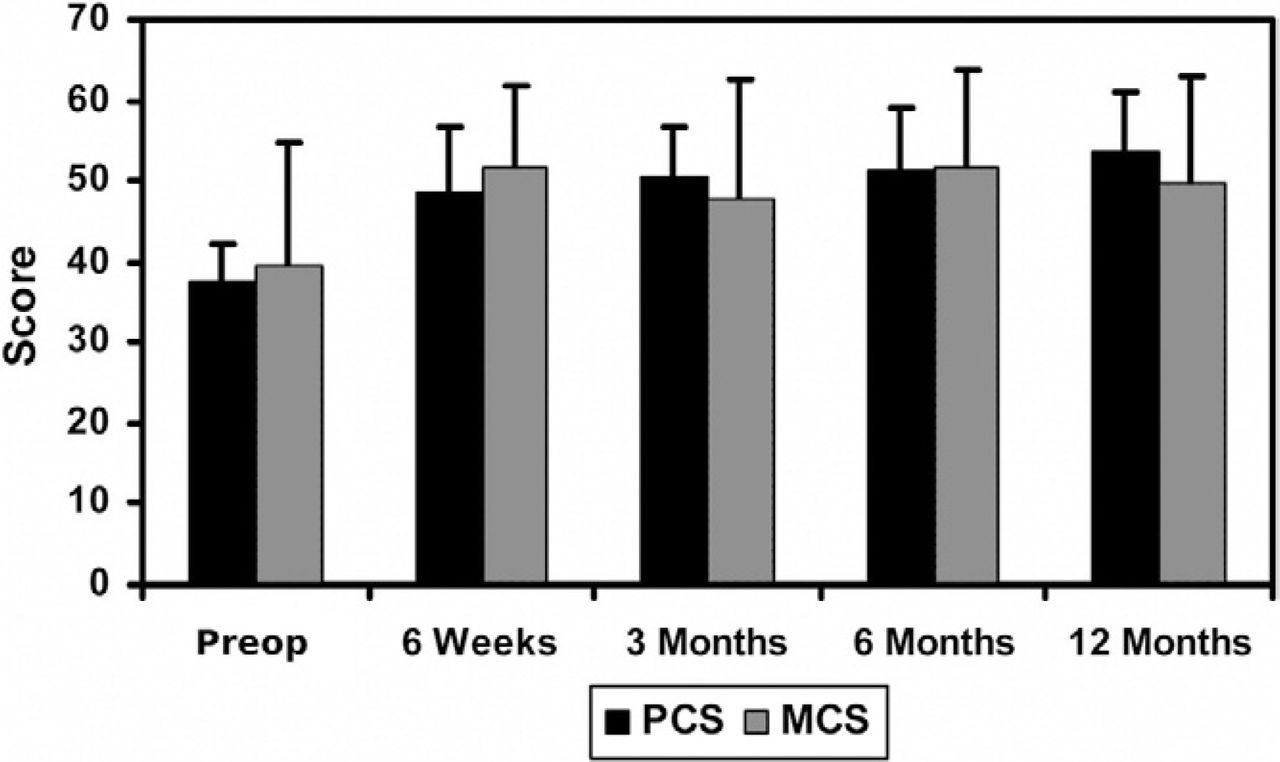
Mean preoperative PCS and MCS scores were 37.7 ± 4.4 and 39.6 ± 15.0, respectively. The mean values of both scores increased by greater than 5.42 points at all follow-up time points when compared to the preoperative values (Fig. 5), indicating that the quality of life of the patients has been improved.

Mean SF-36 scores at all follow up evaluations. Error bars indicate standard deviations.
Patient satisfaction
Ten out of 12 patients (83%) indicated that they would definitely or probably recommend this treatment to others, and 2 patients would not recommend the treatment after 12 months. Overall, all patients were satisfied with their treatment, as shown by an average score of 83.5 ± 26.8 mm on the patient satisfaction scale.
Work status
Prior to surgery, 3 patients (25%) were not working and 7 patients (58%) were able to work but with restrictions. Only 2 patients (17%) were able to work without restrictions. At the 12-month follow-up, all patients were working. Ten patients (83%) were able to work without any restrictions and 2 patients (17%) were working with restrictions.
Radiographic outcomes
Range of motion
The total range of motion at the index level was 12.0° ± 6.2° preoperatively and 13.3° ± 5.5° at the 12-month follow- up. The total range of motion at the adjacent level was 10.8° ± 5.5° preoperatively and 13.3° ± 5.0° at the 12- month follow-up. The range of motion has been maintained from the baseline values (Table 3). This range of motion is within the normal values previously reported.19
Range of motion pre-operatively and at the 12 month follow up evaluation for operative and adjacent levels
Disc height
Disc height (DH) was measured preoperatively and immediately postoperatively (baseline) for each level implanted (16 levels).
The change in disc height (ΔDH) between the pre-op and baseline values and the change between the baseline and last follow-up was calculated (results listed in Table 4). The disc height was restored and maintained to within 1 mm through the last follow-up time point.
Change in disc height
Subsidence, migration, expulsion, loosening
There was no incidence of subsidence, migration, or expulsion for any of the implanted devices.
Fourteen of the 16 implanted devices exhibited no radiolucent lines along the device/implant interface. Mild radiolucency was observed at the 3- and 6-month follow-ups for 2 patients with a 2-level implantation. In both cases, radiolucency was noted at the L4-5 level. At 12 months, this mild radiolucency was only observed in 1 of these patients. Additionally, at 12 months, moderate radiolucency was noted at the L4-L5 level of another patient with a 2-level implantation.
Single versus 2-level implantation
At the 12-month follow-up time point, VAS low back pain scores were 8.9 ± 7.6 and 31.8 ± 41.4 for the single and 2-level implantation groups, respectively. ODI scores were 8.0 ± 8.6 and 22.0 ± 21.3 for the single and 2-level implantation groups, respectively. These results were not statistically significant for either outcome.
Comparison to other devices
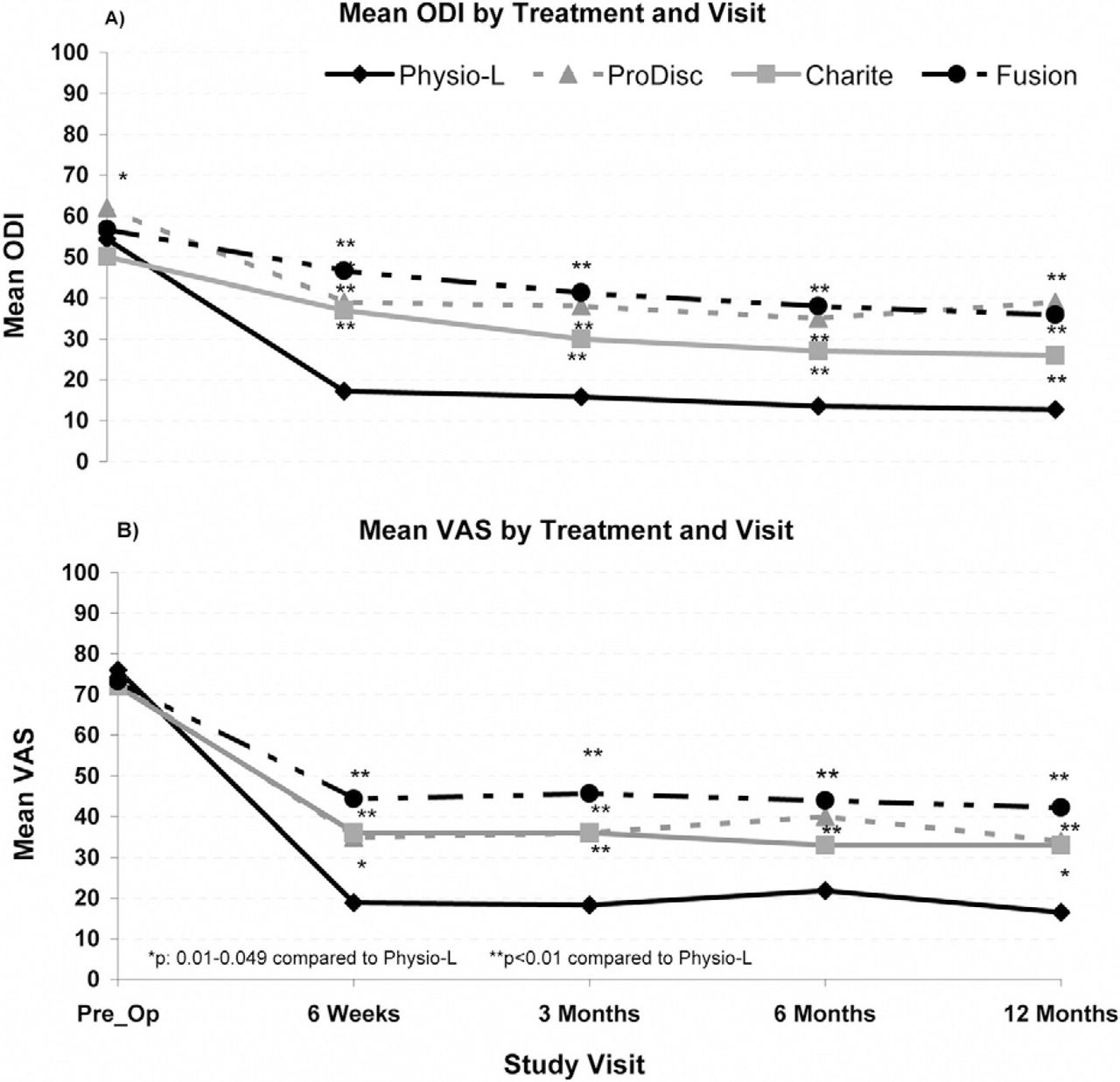
For all treatment modalities (Physio-L, Charite, Prodisc- L and fusion), a minimal clinically important difference (MCID) was noted between the preoperative and postoperative ODI and VAS scores at all follow-up time points. Moreover, compared to all other treatments, the current device showed a statistically significant and clinically relevant improvement in both ODI and VAS scores at all follow-up time points when compared to the fusion and ProDisc-L treatment modalities (P < .05). A statistically significant difference was seen between the current device and the Charite at 6 weeks and 3 and 12 months (Fig. 6). There was no significant difference in preoperative scores between the current device and any other treatment modality.
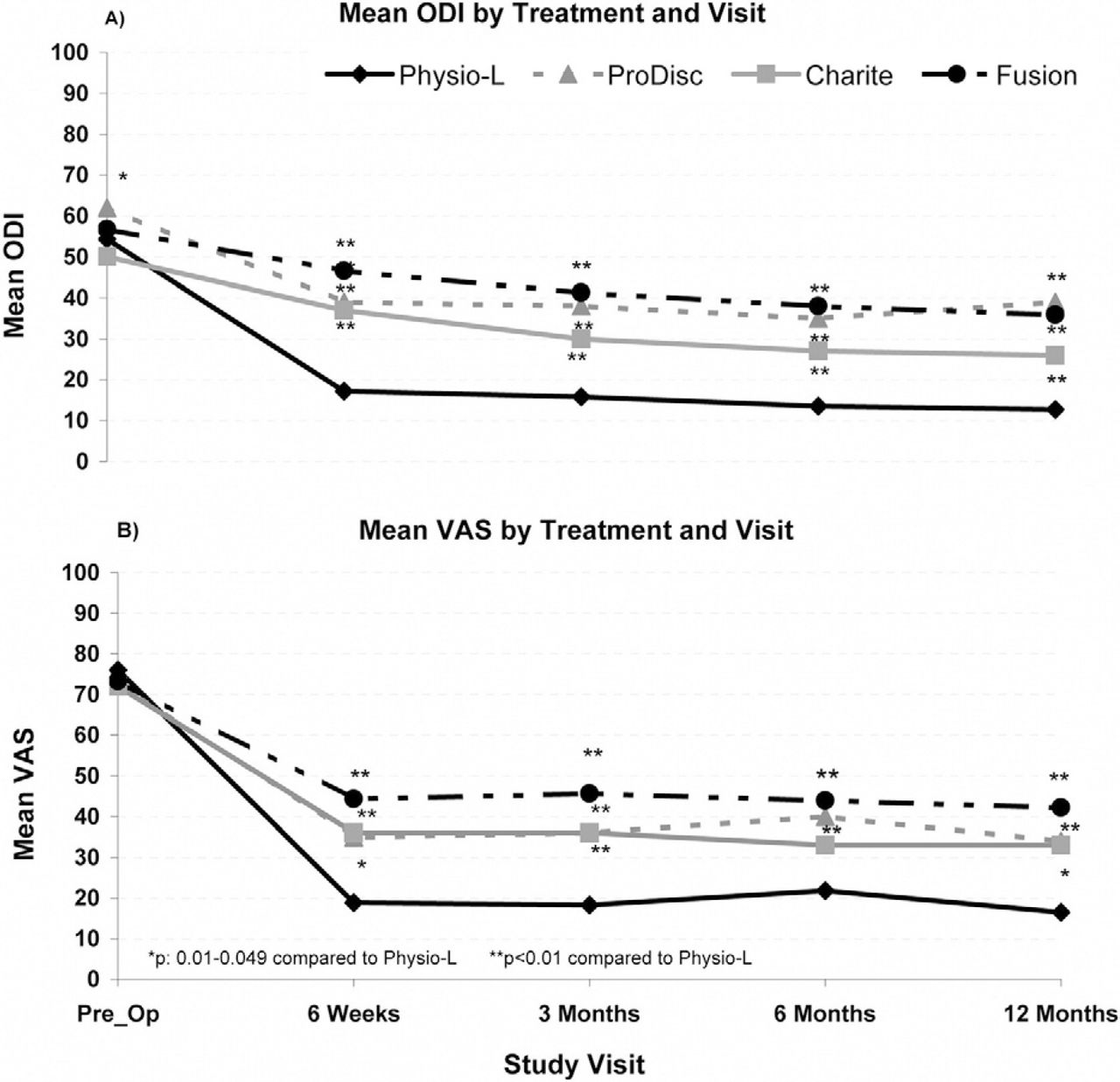
Comparison of the clinical outcomes for the Physio-L versus the Charite, ProDisc-L, and Fusion for A) ODI scores and, B) VAS low back pain scores.
Discussion
This is the first report on an elastomeric lumbar artificial disc using a new generation high endurance polyurethane material system, and shows that, at least after 12 months, the devices have remained intact and are performing as intended. Furthermore, the patients have improved pain relief and function, and all have returned to work.
The present investigation employed a clinical protocol similar to those of previous category 1 studies and included validated clinical outcome measures.16–19 Consequently, it is possible to compare the current data with that previously reported for the Charite and ProDisc-L discs as well as their fusion controls. These short-term results and clinical comparisons are encouraging and warrant further investigation of the device in a larger study with longer-term follow-up results.
Short-term clinical results appear to be predictive of long-term outcomes in similar studies. A retrospective analysis using the Charite and ProDisc-L IDE studies was performed to compare the 6 month mean VAS low back pain and ODI outcomes measures to the same measures at 12, 24, and 60 months.16, 17, 19 No statistically significant difference was found between the 6-month follow-up time point and the 12-, 24-, or 60-month follow-up time point for either the VAS or ODI.
The superior results for the current device, compared to the Charite, ProDisc-L, and fusion, may be explained by the more physiologic compliance of the elastomeric core. The concept of disc prostheses made from elastomeric materials capable of providing physiologic levels of shock absorption and resistance to motion has been in development almost as long as the development of the current generation “sliding bearing” disc prostheses.20 It is postulated that these types of discs may reduce or eliminate the concerns expressed over sliding bearing disc designs, including the fixed center of rotation and increased facet joint loading.21, 22 Historically, however, the availability of fatigue-resistant and biostable materials and the method of attaching the elastomer to a metal endplate were difficult obstacles to the design of a successful device.
Early designs of elastomeric disc prostheses resulted in many failures. The C-Flex material used by Lee et al lacked bio-stability resulting in environmental stress cracking and fatigue failure.20 Two separate clinical trials were conducted to assess the safety of the Acroflex elastomeric artificial disc design (Acromed Corporation, Cleveland, OH). This disc was manufactured using a polyolefin rubber core material. Prior to initiating the clinical studies, extensive biocompatibility and biomechanical tests were conducted on the polyolefin core material including cytotoxicity, compressive creep, peel strength at the titanium-rubber interface and compression, torsion, and shear endurance testing.12, 23, 24 The Acroflex disc was implanted into 28 patients at 1 or 2 levels between L4-S1.11, 12 Ten patients (36%) experienced tears in the polyolefin material, a condition which leads to revision surgery in many affected patients between 2 and 4 years following surgery. The most common mode of failure was anterior-inferior peripheral tears in the rubber that are characteristic of material failure during flexion-extension bending. The authors found no reference that describes any bending endurance testing that was conducted in the laboratory prior to the clinical trial.
More recently, a different family of polymers, polycarbonate polyurethanes, has become available, with longer fatigue life and high resistance to environmental stress cracking.25–28 Notably, polycarbonate polyurethane has been incorporated successfully in various load bearing nonfusion spinal devices such as the Bryan cervical disc (Medtronic, Memphis, TN), Dynesys posterior dynamic fixation (Zimmer, Minneapolis, MN), and the M6 cervical disc prosthesis (Spinal Kinetics, Sunnyvale, CA). The current device also employs such a polycarbonate polyurethane. Prior to clinical implantation, the current device was endurance- tested at least 10 million cycles in multiple modes of testing including flexion-extension, lateral bending, and combined bending to ensure that the previously observed types of failures did not occur.29
Conclusion
That the current device is safe and effective has been demonstrated by improved pain relief and functional recovery without any implant failures, significant device related complications, or adverse incidents at the 12-month follow- up time point. The clinical results for VAS and ODI are superior to other marketed artificial lumbar discs at the same follow-up timeframes. It is postulated that this clinical difference may be due to the compliant nature of the elastomer used in this new generation of elastomeric discs.
Extended references
A prospective, randomized, multicenter Food and Drug Administration investigational device exemptions study of lumbar total disc replacement with the CHARITE artificial disc versus lumbar fusion: part I: evaluation of clinical outcomes.
Blumenthal S, McAfee PC, Guyer RD, et al.
STUDY DESIGN: A prospective, randomized, multicenter, Food and Drug Administration-regulated Investigational Device Exemption clinical trial. OBJECTIVES: The purpose of this study was to compare the safety and effectiveness of lumbar total disc replacement, using the CHARITE artificial disc (DePuy Spine, Raynham, MA), with anterior lumbar interbody fusion, for the treatment of single-level degenerative disc disease from L4-S1 unresponsive to nonoperative treatment. SUMMARY OF BACKGROUND DATA: Reported results of lumbar total disc replacement have been favorable, but studies have been limited to retrospective case series and/or small sample sizes. METHODS: Three hundred four (304) patients were enrolled in the study at 14 centers across the United States and randomized in a 2:1 ratio to treatment with the CHARITE artificial disc or the control group, instrumented anterior lumbar interbody fusion. Data were collected pre- and perioperatively at 6 weeks and at 3, 6, 12, and 24 months following surgery. The key clinical outcome measures were a Visual Analog Scale assessing back pain, the Oswestry Disability Index questionnaire, and the SF-36 Health Survey. RESULTS: Patients in both groups improved significantly following surgery. Patients in the CHARITE artificial disc group recovered faster than patients in the control group. Patients in the CHARITE artificial disc group had lower levels of disability at every time interval from 6 weeks to 24 months, compared with the control group, with statistically lower pain and disability scores at all but the 24 month follow-up (P < 0.05). At the 24-month follow-up period, a significantly greater percentage of patients in the CHARITE artificial disc group expressed satisfaction with their treatment and would have the same treatment again, compared with the fusion group (P < 0.05). The hospital stay was significantly shorter in the CHARITE artificial disc group (P < 0.05). The complication rate was similar between both groups. CONCLUSIONS: This prospective, randomized, multicenter study demonstrated that quantitative clinical outcome measures following lumbar total disc replacement with the CHARITE artificial disc are at least equivalent to clinical outcomes with anterior lumbar interbody fusion. These results support earlier reports in the literature that total disc replacement with the CHARITE artificial disc is a safe and effective alternative to fusion for the surgical treatment of symptomatic disc degeneration in properly indicated patients. The CHARITE artificial disc group demonstrated statistically significant superiority in two major economic areas, a 1-day shorter hospitalization, and a lower rate of reoperations (5.4% compared with 9.1%). At 24 months, the investigational group had a significantly higher rate of satisfaction (73.7%) than the 53.1% rate of satisfaction in the control group (P = 0.0011). This prospective randomized multicenter study also demonstrated an increase in employment of 9.1% in the investigational group and 7.2% in the control group.
Prospective, randomized, multicenter Food and Drug Administration investigational device exemption study of lumbar total disc replacement with the CHARITE artificial disc versus lumbar fusion: five-year follow-up.
Guyer RD, McAfee PC, Banco RJ, et al.
BACKGROUND CONTEXT: The CHARITE artificial disc, a lumbar spinal arthroplasty device, was approved by the United States Food and Drug Administration in 2004 based on two-year safety and effectiveness data from a multicenter, prospective, randomized investigational device exemption (IDE) study. No long-term, randomized, prospective study on the CHARITE disc or any other artificial disc has been published to date. PURPOSE: The purpose of this study was to compare the safety and effectiveness at the five-year follow-up time point of lumbar total disc replacement using the CHARITE artificial disc (DePuy Spine, Raynham, MA) with that of anterior lumbar interbody fusion (ALIF) with BAK cages and iliac crest autograft, for the treatment of single-level degenerative disc disease from L4 to S1, unresponsive to nonoperative treatment. STUDY DESIGN/SETTING: Randomized controlled trial-five-year follow-up. PATIENT SAMPLE: Ninety CHARITE patients and 43 BAK patients. OUTCOME MEASURES: Self-reported measures: visual analog scale (VAS); validated Oswestry disability index (ODI version 1.0); Short-Form 36 Questionnaire, and patient satisfaction. Physiologic measures: radiographic range of motion, disc height, and segmental translation. Functional measures: work status. METHODS: Of the 375 subjects enrolled in the CHARITE IDE trial, 277 were eligible for the five-year study and 160 patients thereof completed the five-year follow-up. The completers included 133 randomized patients. Overall success was defined as improvement> or =15 pts in ODI vs. baseline, no device failure, absence of major complications, and maintenance or improvement of neurological status. Additional clinical outcomes included an ODI questionnaire as well as VAS, SF-36, and patient satisfaction surveys. Work status was tracked for all patients. Safety assessments included occurrence and severity of adverse events and device failures. Radiographic analyses such as index- and adjacent-level range of motion, segmental translation, disc height, and longitudinal ossification were also carried out. RESULTS: Overall success was 57.8% in the CHARITE group vs. 51.2% in the BAK group (Blackwelder's test: p = 0.0359, Delta = 0.10). In addition, mean changes from baseline for ODI (CHARITE: −24.0 pts vs. BAK: −27.5 pts), VAS pain scores (CHARITE: −38.7 vs. BAK: −40.0), and SF-36 questionnaires (SF-36 Physical Component Scores [PCS]: CHARITE: 12.6 pts vs. BAK: 12.3 pts) were similar across groups. In patient satisfaction surveys, 78% of CHARITE patients were satisfied vs. 72% of BAK patients. A total of 65.6% patients in the CHARITE group vs. 46.5% patients in the BAK group were employed full-time. This difference was statistically significant (p = 0.0403). Long-term disability was recorded for 8.0% of CHARITE patients and 20.9% of BAK patients, a difference that was also statistically significant (p = 0.0441). Additional indexlevel surgery was performed in 7.7% of CHARITE patients and 16.3% of BAK patients. Radiographic findings included operative and adjacent-level range of motion (ROM), intervertebral disc height and segmental translation. At the fiveyear follow-up, the mean ROM at the index level was 6.0 degrees for CHARITE patients and 1.0 degrees for BAK patients. Changes in disc height were also similar for both CHARITE and BAK patients (0.7 mm for both groups, p = 0.9827). Segmental translation was 0.4 and 0.8mm in patients implanted with CHARITE at L4-L5 vs. L5-S1, respectively, and 0.1mm in BAK patients. CONCLUSIONS: The results of this five-year, prospective, randomized multicenter study are consistent with the two-year reports of noninferiority of CHARITE artificial disc vs. ALIF with BAK and iliac crest autograft. No statistical differences were found in clinical outcomes between groups. In addition, CHARITE patients reached a statistically greater rate of part- and full-time employment and a statistically lower rate of long-term disability, compared with BAK patients. Radiographically, the ROMs at indexand adjacent levels were not statistically different from those observed at two-years postsurgery.
A prospective, randomized, multicenter Food and Drug Administration investigational device exemption study of lumbar total disc replacement with the CHARITE artificial disc versus lumbar fusion: part II: evaluation of radiographic outcomes and correlation of surgical technique accuracy with clinical outcomes.
McAfee PC, Cunningham B, Holsapple G, et al.
STUDY DESIGN: A prospective, randomized, multicenter, Food and Drug Administration-regulated, investigational device exemption clinical trial. OBJECTIVES: To compare the safety and effectiveness of lumbar total disc replacement (TDR) with the CHARITE artificial disc (DePuy Spine, Raynham, MA) to anterior lumbar interbody fusion for the treatment of single-level degenerative disc disease from L4-S1 unresponsive to nonoperative treatment. In addition, to evaluate the radiographic outcomes of lumbar artificial disc replacement at either L4-L5 or L5-S1 with the CHARITE artificial disc as compared to anterior lumbar interbody fusion with cylindrical cages and iliac crest bone graft; and to determine if a correlation exists between clinical outcomes and surgical accuracy of TDR placement within the disc space. SUMMARY OF BACKGROUND DATA: Prior investigators have reported excellent radiographic results with the CHARITE artificial disc for the treatment of lumbar degenerative disc disease. These encouraging results are the product of retrospective reviews without a control. Very few studies have reported on the segmental motion of an intervertebral level implanted with an artificial disc, and no studies have reported a correlation of radiographic and clinical outcomes. METHODS: A prospective, randomized, multicenter, US Food and Drug Administration, investigational device exemption study with 24-month follow-up was performed at 14 centers throughout the United States. A total of 304 subjects were randomized in a 2:1 ratio, with 205 in the investigational group (TDR with the CHARITE artificial disc) and 99 in the control group (anterior lumbar interbody fusion with BAK cages and iliac crest bone graft). A total of 71 TDR training cases were performed (up to 5 at each site) before randomization beginning at each site. Plain radiographs were analyzed for each subject in both groups regarding range of motion (ROM) in flexion/extension, restoration of disc space height, and subsidence. Prosthesis placement in the coronal and midsagittal planes was analyzed for the 276 patients with TDR. Correlations were performed between prosthesis placement and clinical outcomes. RESULTS: Patients in the investigational group had a 13.6% mean increase, and those in the control group an 82.5% decrease in mean flexion/extension ROM at 24 months postoperatively compared to baseline. Patients in the investigational group had significantly better restoration of disc height than the control group (P < 0.05). There was significantly less subsidence in the investigational group compared to the control group (P < 0.05). The surgical technical accuracy of CHARITE artificial disc placement was divided into 3 groups: I, ideal (83%); II, suboptimal (11%); and III, poor (6%), and correlated with clinical outcomes. The flexion/ extension ROM and prosthesis function improved with the surgical technical accuracy of radiographic placement (P = 0.003). CONCLUSIONS: Preoperative ROM in flexion/ extension was restored and maintained in patients receiving a TDR. TDR with the CHARITE artificial disc resulted in significantly better restoration of disc space height, and significantly less subsidence than anterior interbody fusion with BAK cages. Clinical outcomes and flexion/extension ROM correlated with surgical technical accuracy of CHARITE artificial disc placement. In the majority of cases, placement of the CHARITE artificial disc was ideal.
Results of the prospective, randomized, multicenter Food and Drug Administration investigational device exemption study of the Pro- Disc-L total disc replacement versus circumferential fusion for the treatment of 1-level degenerative disc disease.
Zigler J, Delamarter R, Spivak JM, et al.
STUDY DESIGN: A prospective, randomized, multicenter, Food and Drug Administration-regulated Investigational Device Exemption clinical trial. OBJECTIVE: To evaluate the safety and effectiveness of the ProDisc-L (Synthes Spine, West Chester, PA) lumbar total disc replacement compared to circumferential spinal fusion for the treatment of discogenic pain at 1 vertebral level between L3 and S1. SUMMARY OF BACKGROUND DATA: As part of the Investigational Device Exemption clinical trial, favorable single center results of lumbar total disc replacement with the ProDisc-L have been reported previously. METHODS: Two hundred eighty-six (286) patients were treated on protocol. Patients were evaluated before and after surgery, at 6 weeks, 3, 6, 12, 18, and 24 months. Evaluation at each visit included patient self-assessments, physical and neurologic examinations, and radiographic evaluation. RESULTS: Safety of ProDisc-L implantation was demonstrated with 0% major complications. At 24 months, 91.8% of investigational and 84.5% of control patients reported improvement in the Oswestry Low Back Pain Disability Questionnaire (Oswestry Disability Index [ODI]) from preoperative levels, and 77.2% of investigational and 64.8% of control patients met the > or =15% Oswestry Disability Index improvement criteria. Overall neurologic success in the investigational group was superior to the control group (91.2% investigational and 81.4% control; P = 0.0341). At 6 weeks and 3 months follow-up time points, the ProDisc-L patients recorded SF-36 Health Survey scores significantly higher than the control group (P = 0.018, P = 0.0036, respectively). The visual analog scale pain assessment showed statistically significant improvement from preoperative levels regardless of treatment (P < 0.0001). Visual analog scale patient satisfaction at 24 months showed a statistically significant difference favoring investigational patients over the control group (P = 0.015). Radiographic range of motion was maintained within a normal functional range in 93.7% of investigational patients and averaged 7.7 degrees. CONCLUSIONS: ProDisc-L has been found to be safe and efficacious. In properly chosen patients, ProDisc-L has been shown to be superior to circumferential fusion by multiple clinical criteria.
The implications of constraint in lumbar total disc replacement.
Huang RC, Girardi FP, Cammisa FP Jr, et al.
Lumbar total disc replacement (TDR) is an evolving technique that has the potential to replace arthrodesis as the gold standard surgical treatment of degenerative disc disease. The interaction between host anatomy and physiology and the biomechanical properties of TDR implants will determine the quality of long-term clinical results. However, there is scant literature addressing this subject. The purpose of this article is to discuss the implications of biomechanical constraint in TDR. Based upon available data for normal motion segments and the design of two TDRs currently in clinical trials, unconstrained designs appear to have a kinematic advantage. They are more likely to provide a physiologic mobile instantaneous axis of rotation (IAR), which may explain why they display greater range of motion in vivo. Their lack of constraint may prevent excessive facet joint or capsuloligamentous loads in the extremes of flexion and extension. Furthermore, since the IAR is mobile, they may be less sensitive to small errors in implant placement. On the other hand, constrained devices appear to have an advantage in protection of the posterior elements from shear loading. Spinal shear loads of considerable magnitude occur during activities of daily living. Whether the transference of stresses to the implant and implant-bone interface is clinically significant is unknown. Although this article focuses on two specific TDR designs, future designs will need to account for the same kinematic and loading concerns regarding constraint. We hope this discussion will assist clinicians and researchers in the design, selection, and clinical comparison of present and future TDR implants.
- © 2010 SAS - The International Society for the Advancement of Spine Surgery. Published by Elsevier Inc. All rights reserved.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.