


ABSTRACT
Background: Many early cervical total disc replacements (TDRs) produced motion through a ball-and-socket action, with metal endplates articulating with a plastic core. Polyetheretherketone (PEEK) is used increasingly for spinal implants due to its mechanical properties and lack of artifacts on imaging. A TDR was designed with titanium-coated PEEK endplates and a ceramic core. The purpose of this study was to compare this TDR with anterior cervical discectomy and fusion (ACDF) to treat single-level cervical disc degeneration.
Methods: This was a prospective, nonrandomized, historically controlled, multicenter US Food and Drug Administration (FDA) Investigational Device Exemption (IDE) trial. Patients received the PEEK-on-ceramic Simplify® Cervical Artificial Disc (n = 150). The historic control group included 117 propensity-matched ACDF patients from an earlier IDE trial. The primary outcome was a composite success classification at the 24-month follow-up. Outcome measures included the Neck Disability Index (NDI), neurological status, adverse events, subsequent surgery, a visual analog scale assessing neck and arm pain, and the Dysphagia Handicap Index. Radiographic assessment included flexion/extension range of motion and heterotopic ossification. Facet joints were assessed at 24 months using MRI.
Results: The success rate was significantly greater in the TDR group vs the ACDF group (93.0% vs 73.6%; P < .001). Mean NDI, neck pain, and arm pain scores improved significantly in both groups at all follow-up points. Mean NDI scores in the TDR group were significantly lower than ACDF scores at all follow-up points. There were no significant differences in the rates of serious adverse events. The range of motion of the TDR level had increased significantly by 3 months and remained so throughout follow-up. Facet joint assessment by MRI in the TDR group showed little change from preoperation.
Conclusions: The TDR had an acceptable safety profile and a significantly greater composite success rate than ACDF. These results support that the PEEK-on-ceramic TDR is a viable alternative to ACDF for single-level symptomatic disc degeneration.
Clinical Relevance: This study found that the PEEK-on-ceramic TDR is a viable treatment for symptoms related to cervical disc degeneration and offers similar or superior outcomes compared with fusion.
Level of Evidence: 2.
INTRODUCTION
There have been multiple prospective, randomized, controlled trials comparing cervical total disc replacement (TDR) and anterior cervical discectomy and fusion (ACDF). Multiple meta-analyses of these and other studies have found that TDR produces outcomes superior to ACDF.1–3 The first cervical TDRs used on a large-scale basis produced motion through a ball-and-socket or mobile-bearing action, with metal endplates articulating with a plastic core. Various designs followed including metal-on-metal devices and a titanium-ceramic composite device, as well as a compressible disc with metal endplates. The use of polyetheretherketone (PEEK) material for spinal implants continues to increase, primarily due to its mechanical properties and lack of producing artifacts on imaging studies.4,5 Though less extensive than cervical fusion cages, there has been investigation into the use of PEEK for cervical TDR.6–12 Despite general optimism, there was concern about long-term degradation on articulating PEEK-on-PEEK surfaces.10 This may be particularly problematic and cause uneven loading if the implant is not ideally positioned. Another concern with PEEK is the bioinert surface. However, this has been addressed in many implants by adding a textured porous coating, often titanium, a material with favorable osseointegrative properties.5,13,14 A cervical TDR was designed with commercially pure, titanium-coated PEEK endplates and a biconvex zirconia-toughened alumina ceramic core. This ceramic material has long been used in orthopedic applications, particularly hip implants.15 Biomechanical analysis of the PEEK-on-ceramic device found an acceptable wear profile with no evidence of runaway wear, endplate perforation, or component fracture in idealized or aggressive wear simulation testing.8 The purpose of the study was to compare the PEEK-on-ceramic TDR to ACDF for the treatment of cervical disc degeneration at a single level.
METHODS
Device
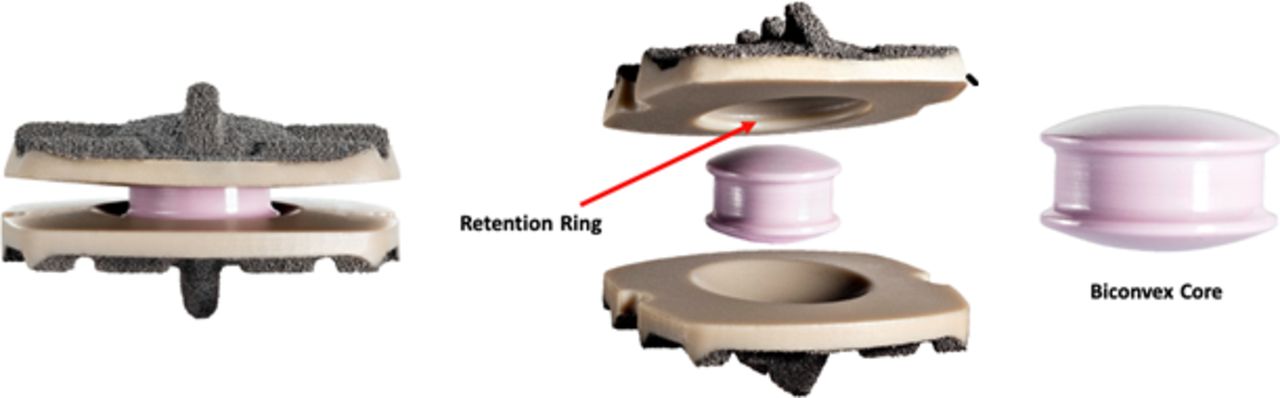
The Simplify® Cervical Artificial Disc (Nuvasive, Inc.; San Diego, CA) comprises PEEK endplates and a mobile zirconia-toughened alumina ceramic core (Figure 1). The endplates have a commercially pure titanium coating. The articulating surfaces on the endplates have a concave surface, and the core has two convex surfaces. The superior and inferior endplates are available in 3 footprints (small, medium, large), 3 thicknesses resulting in 3 device heights (4, 5, and 6 mm), and 2 lordosis angles, 0° and 5°. The superior endplate has a retention ring feature to retain the core within the endplates. The 4-mm device, which became available after approximately 13% of patients were enrolled, was used in 39% of study patients, the 5-mm in 53%, and the 6-mm in 8% of patients. With respect to the lordosis of the implants, the 0° implant was used in 85% of patients and the 5° implant in the remaining 15%.

The PEEK-on-ceramic device investigated in the current study. The PEEK endplates are coated with plasma-spray titanium and the core is ceramic (image courtesy of Simplify Medical, with permission).
Patient Selection and Study Design
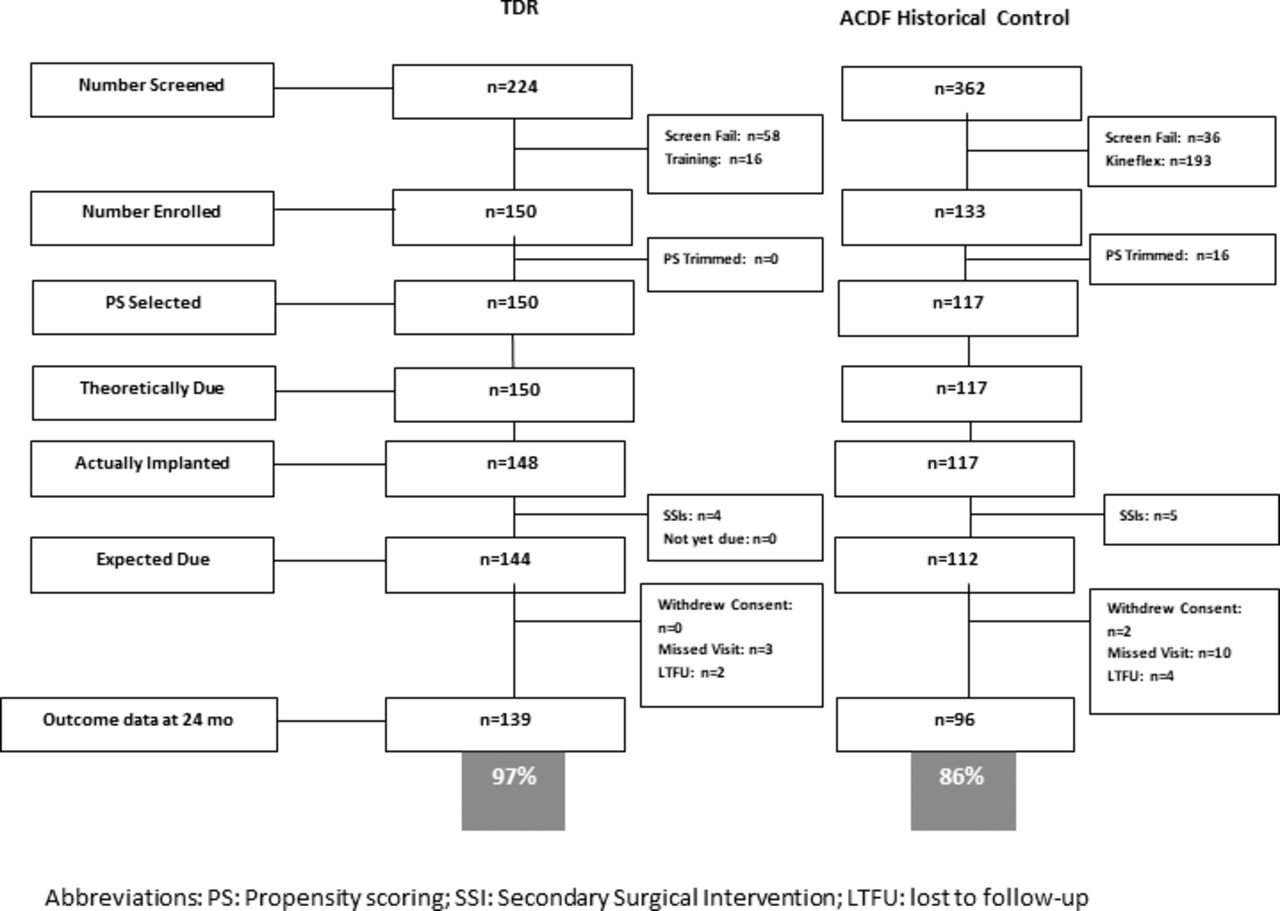
This study was a prospective, nonrandomized, historically controlled, multicenter trial. It was based on the US Food and Drug Administration (FDA) Investigational Device Exemption (IDE) trial for the PEEK-on-ceramic cervical TDR. Data were collected prospectively for 150 study patients from 16 sites (after 16 training cases). Patient selection criteria are provided in Table 1. Outcomes were compared with data collected for 117 historical ACDF control subjects treated at 21 sites as part of a previous IDE TDR trial.16 The inclusion/exclusion criteria used were the same as those used in the current study. At the time of study initiation, several cervical TDR devices were approved for use in the United States. It was thought that this would make it difficult to enroll patients into a randomized study comparing TDR with ACDF. A statistical plan, discussed with and accepted by the FDA, using propensity score (PS) modeling was developed to incorporate the historical ACDF control and to match the baseline covariates to the investigational TDR group. The propensity matching led to the inclusion of all 150 TDR patients enrolled in the trial and 117 of the 133 historical control subjects available for matching (Figure 2). The TDR patients and PS-selected historical control patients had approximately the same multivariate baseline covariate distribution. Table 2 provides an overview of the 2 groups. All patients were treated for single-level symptomatic cervical degenerative disc disease (DDD), defined as intractable cervical radiculopathy with or without neck pain, or with myelopathy. The control group was treated with ACDF using an anterior plate system (DePuy Spine's VG2 allograft bone and Slim-Loc spinal system) and corticocancellous allograft bone.
Inclusion and exclusion criteria.

The derivation of the 2 treatment groups.
Comparison of the TDR and the propensity-matched historical control group. There were no significant differences between the 2 groups.
Outcome Assessment
Patients were evaluated prior to surgery and postoperatively within 2 weeks, 6 weeks, and 3, 6, 12, and 24 months. Patient-completed assessments included the Neck Disability Index (NDI), visual analog scale (VAS) assessing neck and arm pain, quality of life (Short Form-12), Dysphagia Handicap Index (DHI),17 and patient satisfaction. A neurological examination composed of motor, sensory, and reflex assessments was required for the study. Radiographic assessment was performed at each postoperative time point and an MRI was performed at the 24-month follow-up.
The primary study outcome was a clinical composite success (CCS) parameter based on 24-month follow-up values requiring (1) at least a 15-point (of 100) improvement in NDI score from baseline; (2) maintenance/improvement in neurologic status compared with baseline; (3) no device failures; (4) no reoperation, revision, removal, and/or supplemental fixation at the index level, and (5) absence of major adverse events (AEs; defined as definite device relationship that caused permanent neurologic damage related to treated level or below or implant breakage/migration without index level surgery that caused significant dysphagia and/or death).
AE Evaluation
A clinical events committee composed of 3 independent spine surgeons was responsible for the adjudication of AEs (reviewing the relationship to device/procedure, seriousness, severity, determination of major AE, and unanticipated adverse device effects), neurological status, secondary surgical interventions (ie, classification of revision, removal, reoperation, or supplemental fixation), and protocol deviations.
Radiographic and MRI Assessment
All imaging assessments were performed by an independent lab specializing in image analysis (Medical Metrics, Houston, TX). Flexion/extension, neutral anteroposterior, neutral lateral, and lateral bending radiographs were made at required study evaluation points. Radiographic assessment included segmental flexion/extension range of motion (ROM), disc height, and heterotopic ossification (HO). HO was assessed using a 0 to 4 (none to bridging bone) scale commonly used in TDR studies.18
Preoperative and 24-month postoperative MRIs were used to assess the facet joints at the treated level. The facet joints were evaluated using the following classification system.19,20
0. None: normal facet joint space
1. Mild: narrowing of the facet joint space and/or small osteophytes and/or mild hypertrophy of the articular process
2. Moderate: narrowing of the facet joint space and/or moderate osteophytes and/or moderate hypertrophy of the articular process and/or mild subarticular bone erosions
3. Severe: narrowing of the facet joint space and/or large osteophytes and/or sever hypertrophy of the articular process and/or severe subarticular bone erosions and/or subchondral cysts
Data Analysis
Estimated CCS rates for each treatment group, as well as the treatment group differences and 90% confidence intervals (CI) for group differences controlling for PS subclass, were determined using a generalized linear model with results expressed on the probability scale. This was accomplished using the SAS software procedure GENMOD (SAS Institute, Cary, NC). The PS-adjusted success rates and standard errors on the probability scale were pooled to determine the standard error of the estimated group difference. The CCS were adjusted for PS subclass and therefore will not match the unadjusted (x/N) percentages. The lower bound of the 90% CI is equivalent to that of the 1-sided 95% noninferiority CI. The PS model included main effects and important interactions and squared terms for selected baseline variables, such as age and body mass index.
In the study protocol, the null hypothesis was that the probability of achieving CCS for patients implanted with the TDR was no more than .10 smaller than the probability of ACDF control patients achieving CCS. This study would be considered a success if the PS-quintile adjusted, multiple imputation–based, 1-sided P value for rejecting this null hypothesis was less than .05. If the noninferiority study success criterion was met, superiority would be tested. If the PS-quintile adjusted 1-sided P value determined from the multiple imputation was less than .05, it would be concluded that TDR was superior to ACDF.
The primary analysis set includes all TDR patients (n = 150) and controls selected into a PS subclass (n = 117). Of 150 TDR patients, 142 (94.7%) were evaluable for CCS. Among 117 historical controls, 96 (82.1%) were evaluable for primary CCS endpoint. To account for missing data, a fully conditional specification21 approach was used to produce 20 multiply imputed (MI)22 completed data sets on the basis of PS subclass and CCS endpoints determined at intermediate time points as implemented in the SAS software procedure MI. Group comparisons in secondary endpoints also controlled for PS subclass. Within-group descriptive summaries (not adjusted for PS) included means and standard deviations for continuous variables and counts and percentages for categorical variables. Nominal 95% PS-adjusted CIs and P values were reported for selected secondary endpoints. Data were censored following a secondary surgical intervention (SSI) and intraoperative deviation at the index level.
RESULTS
Clinical Outcome
On the primary outcome evaluation, overall composite success based on the 5-point combined success criteria at the 24-month follow-up, the success rate was significantly greater in the TDR group than in the ACDF control group (93.0% vs 73.6%; P < .01 [PS adjusted]; Table 3) and superiority criteria were met. When comparing the success rates on each of the individual parameters included in the composite assessment, the TDR group had significantly greater rates on NDI success and neurological status success. There was not a statistical difference between groups on the assessments of no additional surgery. Statistical assessment between groups was not estimable for no device failures, and no major AEs. The TDR had similar or better success rates on all criteria parameters, compared with ACDF.
Comparison of the overall composite success score and the individual components of the composite success in the TDR group and the control fusion group.
Neck Disability Index
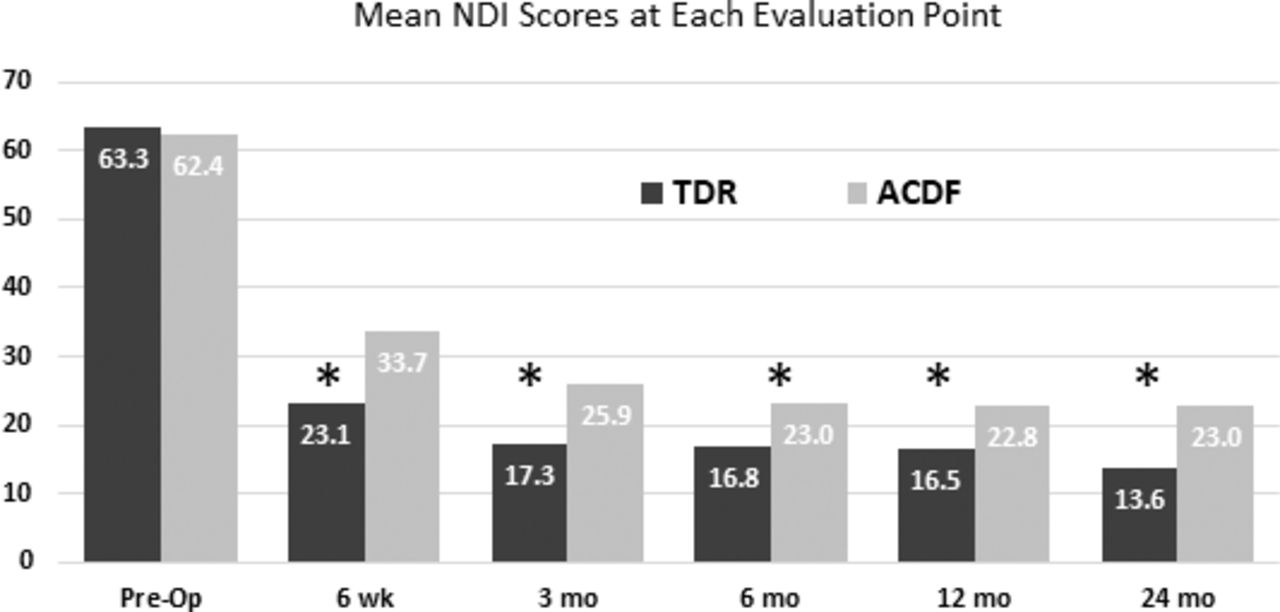
The mean preoperative NDI scores were similar in the TDR and ACDF groups (Figure 3). The mean NDI scores in both groups were improved significantly at all follow-up points (P < .001) The mean NDI scores in the TDR group were significantly less than in the fusion group at all follow-up points (all P < .025). The mean change from baseline at 24 months was −49.4 (P < .001; 95% CI, −46.6 to −52.2) in TDR group and −38.9 (P < .001; 95% CI, −34.7 to −43.1) in the ACDF group. At the 24-month follow-up, significantly more TDR patients achieved the criteria to be classified as reaching clinically relevant improvement on the NDI (≥15-point decrease 97.9% vs 88.0%; P < .05).

The mean Neck Disability Index (NDI) scores were similar in the 2 groups prior to surgery and were significantly improved throughout follow-up in both groups (P < .001), with the total disc replacement (TDR) group having significantly lower scores at all follow-up points (*P < .025; propensity score [PS] adjusted). Subjects were censored at index-level secondary surgical interventions and intraoperative deviations.
Visual Analog Scale
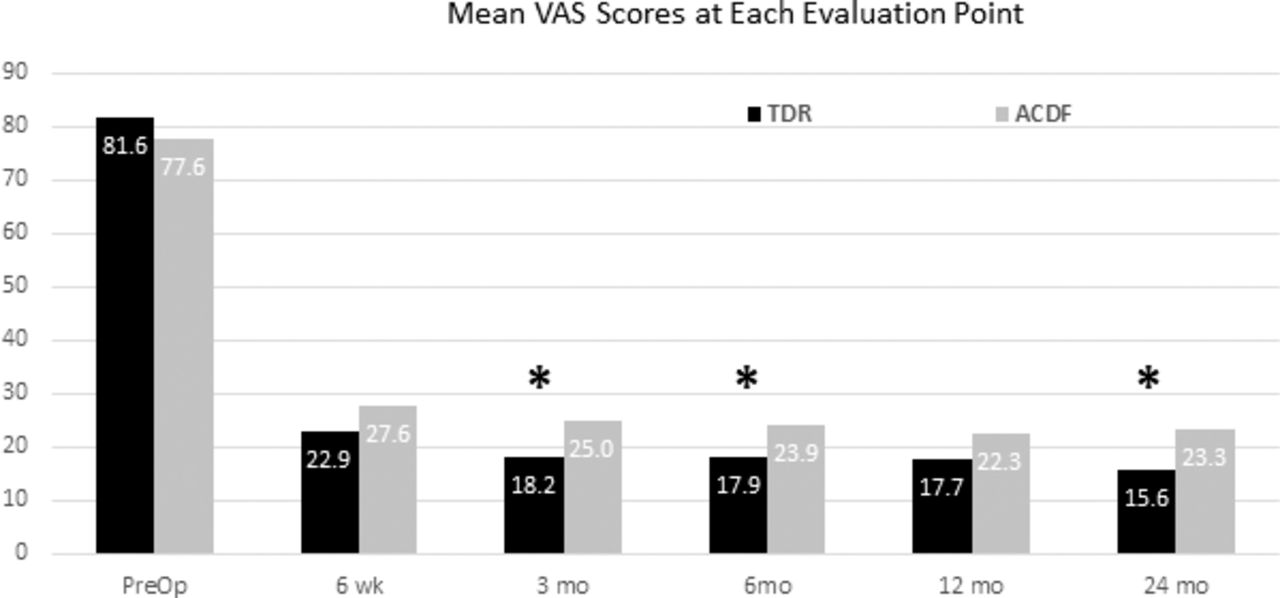
The mean VAS neck and arm pain scores were similar in the TDR and ACDF groups prior to surgery and improved significantly in both groups at all follow-up points (all P < .001; Figure 4). At the 3-, 6-, and 24-month follow-up points, the scores were significantly less in the TDR group (P < .05). The mean change from baseline at 24 months was −66.4 (P < .001; 95% CI, −62.7 to −70.0) in the TDR group and −53.7 (P < .001; 95% CI, −48.3 to −59.1) in the ACDF group.

The mean visual analog scale (VAS) neck and arm pain scores were similar in the 2 groups prior to surgery and were significantly improved at all follow-up points (P < .001). The mean scores in the total disc replacement (TDR) group were statistically significantly less than ACDF values at the 3-, 6-, and 24-month follow-ups (P < .05, propensity score [PS] adjusted). Subjects were censored at index level secondary surgical interventions and intraoperative deviations.
Dysphagia Handicap Index
The mean DHI score in the TDR group was 6.2 prior to surgery and increased to 7.0 at the 6-week follow-up. By 3 months after surgery, the mean value had decreased to 4.2 and remained near this value throughout follow-up. The DHI was not administered in the ACDF historic control group.
Neurological Evaluation
Neurological success was defined as maintenance or improvement in neurologic status at the 24-month follow-up compared with the patient's preoperative state. In the TDR group, only 1 patient was classified as a neurological failure, which was significantly fewer than the 5 failures in the ACDF cohort (0.4% vs 5.9%; P < .02 [PS adjusted]).
Secondary Surgical Interventions
In the TDR group there were no cases of device failure or device migration. Through the 24-month follow-up, there were 4 (2.7%) SSI at the index level in the TDR group and 6 (5.1%) in the ACDF group (1 was not included in overall success and subject accounting because it occurred after the 2-year endpoint). In the TDR group, SSIs included the following: 1 patient underwent TDR removal and had ACDF performed due to recurrent stenosis and worsening degeneration; another patient had an esophageal tear during the surgical approach, which led to a deep wound infection treated by device removal, corpectomy at C7, ACDF C6-T1 and posterior fusion C6-T2; the third patient had symptomatic pseudoarthrosis after undergoing reoperation ACDF at the superior adjacent segment and the TDR level was included in anterior plating; and the fourth patient underwent a decompression at the TDR level to address C7 radiculopathy. In the ACDF group, the 6 SSIs included 2 cases of removal of the anterior plate and insertion of a TDR (not the investigational device; in 1 case due to radiculopathy and pseudoarthrosis in the other); 2 cases of removal of anterior plate and replacement with plating at the index and adjacent segment due to adjacent segment degeneration; and 2 cases of removal of anterior plate and a revised graft and plate (due to graft subsidence in 1 case and pseudoarthrosis in the other).
Adverse Events
Through the 24-month follow-up, the percentage of patients having AEs classified as definitely device related was 0.7% (1/150) in the TDR group and 0.9% (1/117) in the ACDF group. The TDR patient had 2 events related to implant/joint noise and inflammation. In the ACDF group, the event was related to pseudarthrosis. An overview of all the serious AEs is provided in Table 4. There were no statistically significant differences between the TDR and the ACDF control groups.
The percentage of patients with SAEs reported in the 2 study groups.
Radiographic Assessment
Range of Motion
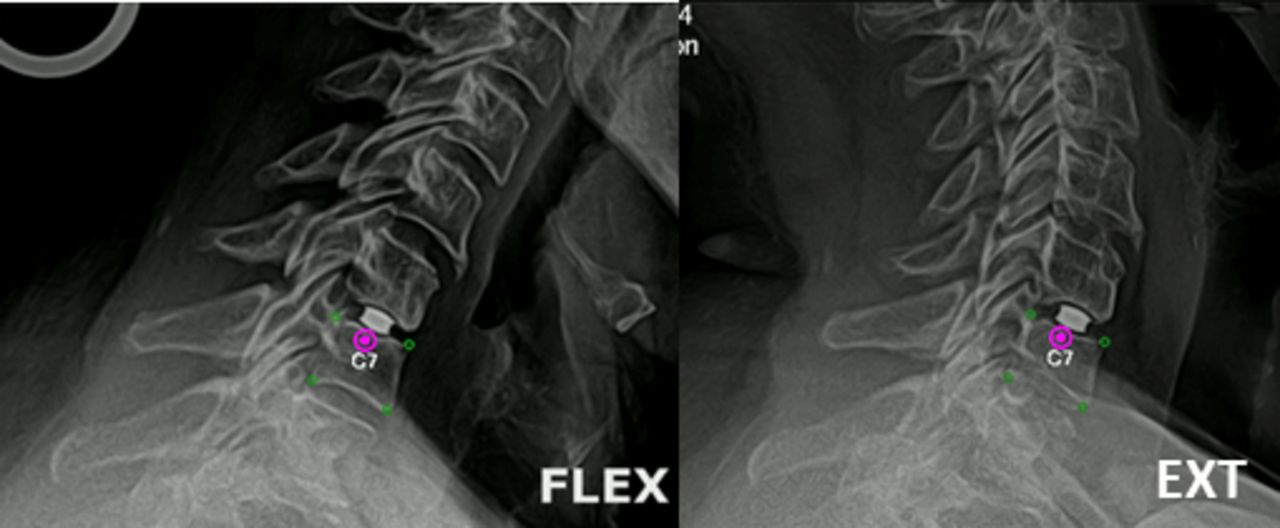
The mean segmental ROM of the treated level in the TDR group was 7.38° prior to surgery. This increased to 8.6° at the 3-month follow-up and continued to improve to 9.6° at the 24-month follow-up (P < .001). Seen in Figure 5 are flexion/extension radiographs showing motion of the implanted level at C6-7 at the 24-month follow-up.

Flexion and extension radiographs from the 24-month follow-up shows the motion of the implant at the C6-7 level.
Heterotopic Ossification
At the 24-month follow-up, 7.2% of TDR patients developed grade 4 (bridging bone) HO (1 patient had grade 3 preoperatively) and an additional 7.9% had grade 3 severe HO (2 patients had grade 2 preoperatively). All of the patients with grade 3 or 4 HO met the 5-point criteria to be classified as having a clinically successful outcome.
Facet Joint Assessment
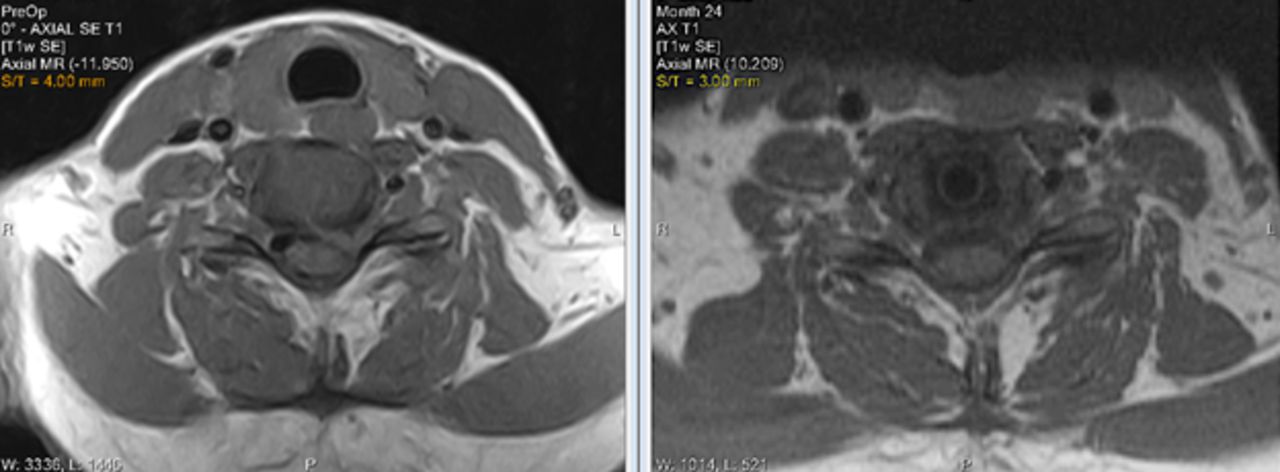
Facet joints at the TDR level were assessed on the basis of MRIs made preoperatively and at 24 months postoperatively. As seen in Figure 6, there was little change in the facet joint degeneration scores. Images are provided in Figure 7 comparing preoperative with postoperative MRIs. Facet joints were not assessed in the ACDF control cohort.

There was little change in the severity of facet joint degeneration at the total disc replacement (TDR) at the 24-month follow-up compared with the preoperative MRIs (subjects censored at index level secondary surgical interventions and intraoperative deviations).
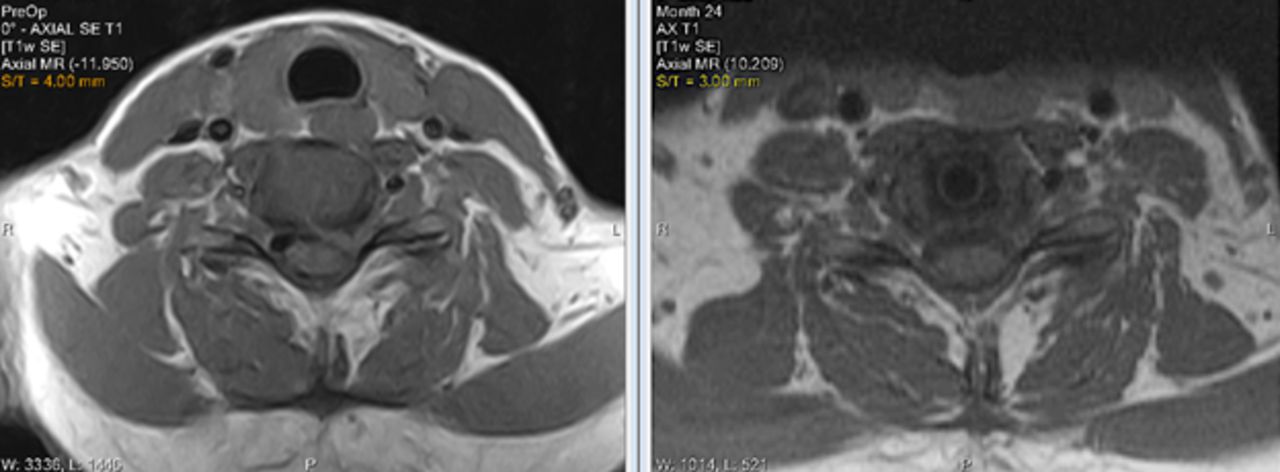
Preoperative and 24-month postoperative MRI of the C6-7 level after total disc replacement (TDR) implantation showing no changes in the facet joints.
DISCUSSION
This prospective IDE trial with a historical propensity-matched ACDF control group found that the PEEK-on-ceramic TDR had an acceptable safety profile and produced clinical outcomes similar or superior to ACDF through the 24-month follow-up. The PS-adjusted composite success rate of 93.0% in the TDR group was significantly greater than that in the ACDF control group (73.6%) and met superiority criteria. It was also numerically greater than results reported23–28 in other FDA-approved single-level cervical TDR studies using similar composite success criteria, with rates ranging from 72.3% to 86.8%. There were no TDR device failures or migrations, and there were no complications related to the PEEK-on-ceramic materials.
Unlike the earliest TDR IDE trials, this study was not randomized. Although generally preferable, it was thought that randomization was not feasible with multiple TDRs already FDA approved for use in the United States. Attempting to randomize to ACDF would place patients in the position of potentially receiving fusion, which has been found to be inferior to TDR in meta-analyses.1,29 This factor may have made it unduly challenging to recruit patients and/or may have biased the study population. Instead, a group of patients randomized to ACDF in a prior TDR vs ACDF study, using the same selection criteria, many of the same protocol elements, and in fact performed by some of the same surgeons as those participating in the current TDR study were propensity matched to the current TDR population. The study plan was discussed with the FDA and found acceptable. This format of using a propensity-matched ACDF historical control group from an earlier randomized trial has also been used in other recent and pending cervical TDR IDE trials.27,30
Several biomechanical studies involving cervical TDR have investigated the potential impact of prosthesis height, particularly with respect to implant sizes that are larger than the natural disc space height. These studies found that increased implant height was associated with decreased ROM31–33 and reduced facet overlap, possibly altering the loading of the adjacent facet joints. These changes could affect the motion of the index level as well as loading of the adjacent segments31,32,34,35 and could also be related to the development of HO.36,37 One study from China found that in 36% of the study population, the height of the cervical disc space was less than the minimal height of available TDRs, which is generally 5 mm.38 In analyzing radiographs from an earlier TDR IDE trial,16 it was noted that 55% of adjacent level discs from C3-4 through C6-7 (n = 261) had an average disc height of less than 4 mm (internal data; Simplify Medical, Sunnyvale, CA), smaller than the minimal prosthesis height available from FDA-approved cervical TDRs. In the current study, 82% of index levels (n = 164) had an average disc height of less than 4 mm (average of anterior disc height and posterior disc height) and a device of 4-mm height was used in 39% of patients.
The rate of grade 3 or 4 HO in the current study was 15.1%. This figure is consistent with rates reported at the 24-month follow-up in the summary of safety and effectiveness data reports submitted to the FDA for other cervical TDR devices applying a similar HO scoring system. In these studies, the rates of HO were 11.3%,39 19.2%,40 15.9%,41 and 4.3%.42 These rates are consistent with a meta-analysis of cervical HO at the 24-month follow-up reporting a pooled rate of grade 3 or grade 4 HO of 16.7% with a 95% confidence interval of 4.6% to 28.9%.43
A potential benefit of the PEEK-on-ceramic materials used in the TDR evaluated in this study is the ability to perform MRI with minimal artifacts compared with that created by devices with metallic components. MRI is generally the imaging modality of choice to evaluate the cervical spine to assess the spinal canal, neuroforamina, intervertebral discs, and facet joints. In the current study, facet joints were assessed using a quantified scoring system, and no significant changes were found at the 24-month follow-up compared with the preoperative images. The lack of artifacts associated with the PEEK-on-ceramic device, which is nickel free, allowed MRI assessment of the spinal canal and exiting nerve roots with little or no distortion at the TDR level. As discussed by Sekhon et al,44 artifacts created by TDRs with metallic endplates, particularly with metals other than titanium, increase the need for computed tomography/myelography to evaluate the cervical spine. This introduces additional risks to the patient as well as costs. The results of this study found that the Simplify® Cervical Artificial Disc produced outcomes similar to or superior on some measures, including superiority in the composite success classification, compared with ACDF, for the treatment of single-level symptomatic cervical disc degeneration. The ROM at the treated level increased from the preoperative value and was maintained throughout follow-up. Safety of the PEEK-on-ceramic disc was established with no device failures in the series. These results support that the device is a viable alternative to ACDF.
ACKNOWLEDGMENTS
The authors would like to acknowledge the following principal investigators for their participation in this trial: Cameron Carmody, MD, and Andrew Park, MD, Texas Spine Consultants, Addison, TX; Kris Radcliff, MD, Rothman Institute, Philadelphia, PA; William Taylor, MD, and Todd Allen, MD, University of California San Diego; Christopher Good, MD, Virginia Spine Institute, Reston, VA; Michael Janssen, DO, Spine Education and Research Foundation, Thornton, CO; Ashvin Patel, MD, Kennedy-White Orthopaedic Center, Sarasota, FL; Andrew Cappuccino, MD, Buffalo Spine Surgery, Lockport, NY; Jon I. White, MD, Orthopaedic Specialty Institute, Irvine, CA; Clint Hill, MD, and K. Brandon Strenge, MD, Orthopaedic Institute of Western Kentucky, and Andy Redmond, MD, Precision Spine Care, Tyler, TX.
Footnotes
Disclosures and COI: Guyer: Consulting: Simplify; Peloza: Speaking and/or Teaching Arrangements: Simplify Medical; Sasso: Research Support (paid directly to institution/employer) Simplify; Coric, Nunley, Musacchio, Bae, and Ohnmeiss: nothing to disclose.
- This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery. Copyright © 2021 ISASS
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.