


Abstract
Background Vertebral compression fractures (VCFs) can cause significant pain and functional impairment, and their cumulative effect can lead to progressive morbidity. This single-arm, prospective feasibility trial, conducted at 4 clinical sites, was undertaken to evaluate the clinical outcomes associated with the use of an innovative vertebral augmentation device, the Kiva VCF Treatment System (Benvenue Medical, Santa Clara, California), in the management of symptomatic VCFs associated with osteoporosis.
Methods Vertebral augmentation treatment was performed for persistent back pain symptoms in 57 patients (mean age, 71.9 ± 10.4 years), including 46 women, with radiologically confirmed VCFs; 36 of these patients (63%) had reached 12 months of follow-up at this data analysis. There were 51 one-level cases, 5 two-level cases, and 1 three-level case, representing 64 treated levels. Back pain severity and condition-specific functional impairment were evaluated with a standard 100-mm visual analog scale and the Oswestry Disability Index (ODI), respectively, before device implantation as well as at 6 weeks, 3 months, and 12 months.
Results Marked clinical improvements were realized in back pain severity and functional impairment through 12 months of follow-up. The mean back pain score on the visual analog scale improved from 79.3 ± 17.2 before treatment to 21.9 ± 21.3, 21.9 ± 24.6, and 23.2 ± 23.3 at 6 weeks, 3 months, and 12 months, respectively. The mean decrease at 12 months was 49.9 ± 30.3 mm, or approximately 66% (P < .0001). Similarly, the mean ODI score improved from 68.1% ± 16.9% before treatment to 27.4% ± 17.2%, 23.8% ± 18.7%, and 23.3% ± 15.5% at 6 weeks, 3 months, and 12 months, respectively, representing a mean change of 39.2 ± 19.6 percentage points, or approximately 63%, at 12 months. Overall clinical success rates based on a 30% improvement in pain severity or greater and maintenance or improvement in the ODI were 91%, 88%, and 89% at 6 weeks, 3 months, and 12 months, respectively. The vertebral augmentation procedure required injection of a mean of 2.2 ± 0.12 mL of cement per vertebral body. There were 5 levels (8%) where cement extravasation was identified radiographically, and none were related to clinical symptoms.
Conclusions These pilot findings are encouraging, suggesting robust and durable clinical improvement after this novel vertebral augmentation procedure in patients with painful VCFs.
Vertebral compression fractures (VCFs) are the earliest clinical manifestation and the most prevalent of the classic fracture types resulting from osteoporosis.1, 2 It is now well understood that the occurrence of vertebral fractures represents a significant negative impact on overall health, where the pain and impaired function associated with the fracture lead to accelerated bone loss, anatomic alterations, and increased risk of subsequent fractures.3, 4
It is estimated that 1.4 million VCFs that cause pain, disability, and diminished quality of life are diagnosed each year worldwide.5 Conservative medical management including analgesic agents, bed rest, physical therapy, and back bracing remains the first line of treatment for medically refractory pain caused by acute, symptomatic VCFs.However, pain resolution can be slow, and symptoms can persist chronically.6 In addition, the annual costs of only medical management of osteoporotic VCFs has been estimated at almost US $14 billion.7
The morbidity and substantial medical costs associated with VCFs have caused a paradigm shift in clinical management toward the goal of more rapid pain relief using percutaneous vertebral augmentation procedures.8 Indeed, the bulk of published evidence suggests that prevalent, symptomatic VCFs can be treated in a minimally invasive fashion, providing noticeable and sustained clinical benefit to afflicted patients.9 In a large (N = 300) randomized controlled trial, Wardlaw et al.10 showed that vertebral augmentation with balloon kyphoplasty in patients with acute, painful VCFs improved quality of life, function, mobility, and pain more rapidly than did nonsurgical management.
This single-arm, prospective feasibility trial was undertaken to evaluate the clinical outcomes and adverse events associated with the use of the Kiva VCF Treatment System (Benvenue Medical, Santa Clara, California) in the treatment of symptomatic VCFs.
Methods
Patients
The data included in this report were collected at 3 clinical sites in Mexico and 1 in Venezuela to prospectively evaluate the preliminary safety and effectiveness of the VCF Treatment System in the treatment of patients sustaining painful VCFs. Specific study eligibility criteria included age at entry of 50 years or greater, 1 to 3 symptomatic VCFs due to osteoporosis, a back pain visual analog scale (VAS) score of 5 or greater, fracture age of less than 6 months, and an Oswestry Disability Index (ODI) score of 30% or greater. Fifty-seven patients with radiologically confirmed VCFs between T6 and L5 underwent vertebral augmentation treatment for persistent back pain symptoms. At this data analysis, 48 patients had reached 6 weeks, 41 had reached 3 months, and 36 had reached 12 months of follow-up. The background characteristics of the overall study group are provided in Table 1. There were 51 single-level cases, 5 two-level cases, and 1 three-level case, representing 64 treated levels. At baseline, patients presented with severe back pain and marked functional impairment.
Patient characteristics (N = 57)
Interventions
Before surgery, all patients underwent a complete physical examination including detailed medical history and complete radiographic imaging studies, including magnetic resonance imaging and thoracolumbar lateral and anteroposterior radiographs, to confirm the presence, location, and severity of VCF.
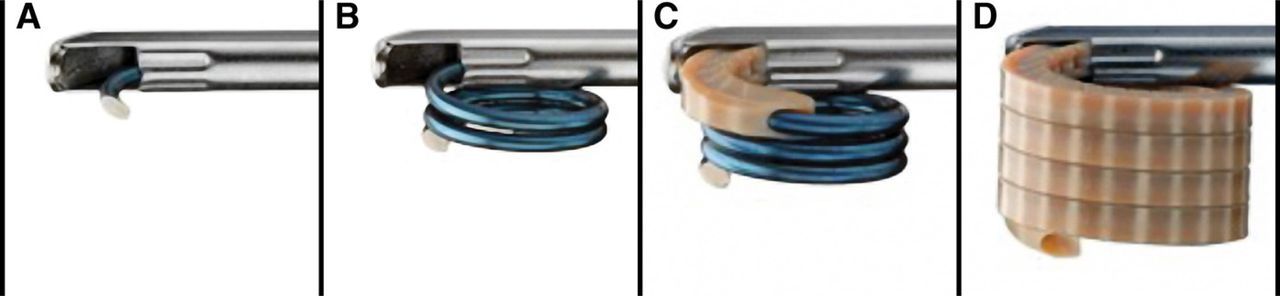
A standard transpedicular percutaneous procedure was used with fluoroscopic guidance to ensure proper needle placement, device deployment, positioning of the implant and injection of bone cement. All patients were treated with the Kiva VCF Treatment System. The VCF Treatment System is a sterile, single-use device consisting of a nitinol Kiva Coil, serving as the access, positioning, and deployment component. This coil is guided through a deployment cannula (Fig. 1) into the cancellous portion of the vertebral body through an external handle mechanism. The coil, like a guidewire, determines the path that the implant will follow. The implant, constructed from PEEK-OPTIMA (Invibio Inc., West Conshohocken, Pennsylvania) with 15% barium sulfate for radiopacity, is delivered over the coil (Fig. 1). As the implant advances over the coil, it may reduce the fracture via height distraction of the vertebral body. The implant is inserted incrementally into the cancellous region of the vertebral body to form a nesting, cylindrical column that provides the desired reduction (Fig. 2). The coil is retracted, leaving the implant in place. Bone cement is injected through the lumen of the implant, which directs the flow of cement to the central part of the vertebral body.

Graphical illustration of VCF Treatment System, consisting of a percutaneously introduced nitinol coil guidewire advanced through a deployment cannula (A) and then fully coiled within the cancellous portion of the fractured vertebral body (B). A radiopaque polyetheretherketone implant is delivered incrementally over the removable coil (C) in a continuous loop to form a nesting, cylindrical column providing vertical displacement that may result in endplate re-elevation and fracture reduction (D).

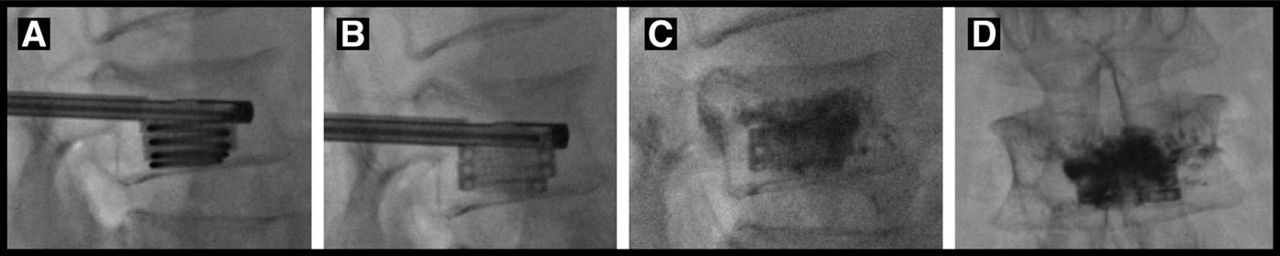
Fluoroscopic images illustrating deployment of the implant into a vertebral body over the removable guidewire in a coiled manner (A). After removal of the coil, the implant is fully deployed (B) to provide structural support to the vertebral body and serve as a conduit for bone cement placement. After bone cement delivery through the lumen of the implant, lateral (C) and anteroposterior (D) fluoroscopic images show contained interdigitation of cement into the adjacent cancellous bone, and the fracture is fully stabilized in situ.
Outcomes
Patient-reported outcomes were measured before device implantation as well as at 6 weeks, 3 months, and 12 months. Back pain severity was evaluated with a standard 100-mm VAS. Condition-specific functional impairment was evaluated with the ODI. Cement extravasation was evaluated from plain radiographs at an independent image analysis core laboratory (Medical Metric Inc., Houston, Texas) by a musculoskeletal radiologist. Newly occurring adjacent and nonadjacent VCFs also were identified by the same radiologist.
Statistical methods
Background characteristics and clinical results are presented as descriptive statistics or frequency and percentage distributions, as appropriate. The degree of clinical improvement in pain and functional outcomes over baseline is displayed graphically by use of line graphs as well as box-and-whisker plots. Baseline values for all outcomes were compared with values at each follow-up interval for statistical significance using the paired t test, 2-tailed. Overall clinical success was defined as a 30% improvement in VAS pain severity or greater and maintenance or improvement in the ODI.11
Results
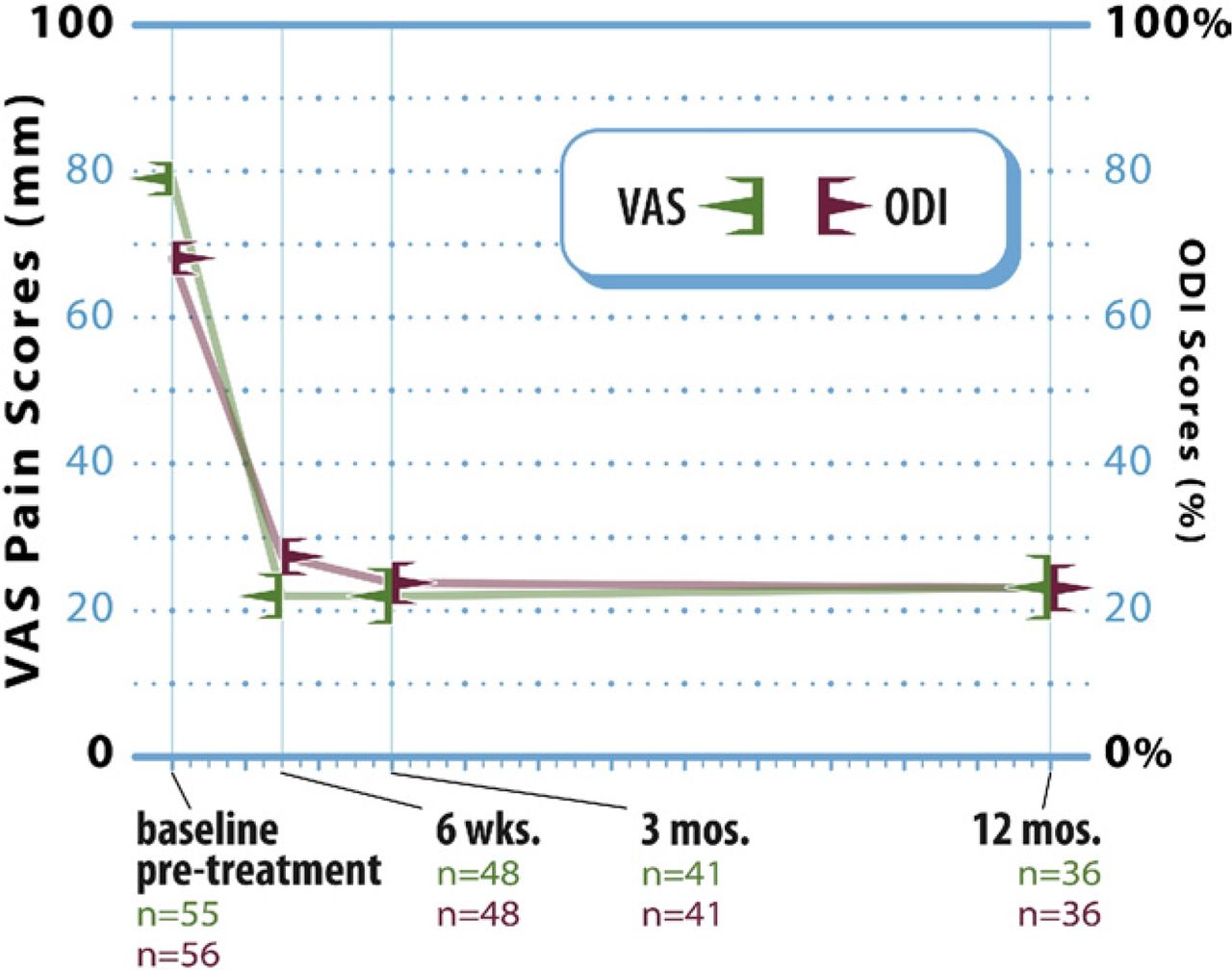
Marked improvement in back pain severity from baseline was realized within 6 weeks of surgery and was sustained through 12 months of postoperative follow-up (Fig. 3). Overall, the mean VAS back pain score improved from 79.3 ± 17.2 before treatment (n = 55) to 21.9 ± 21.3, 21.9 ± 24.6, and 23.2 ± 23.3 at 6 weeks (n = 48), 3 months (n = 41), and 12 months (n = 36), respectively. The mean decrease at 12 months was 49.9 ± 30.3 mm, and the corresponding mean percentage improvement in VAS pain scores was approximately 66%. Compared with before treatment, the degree of pain relief realized at each follow- up interval was statistically significant (P < .0001 for each comparison).
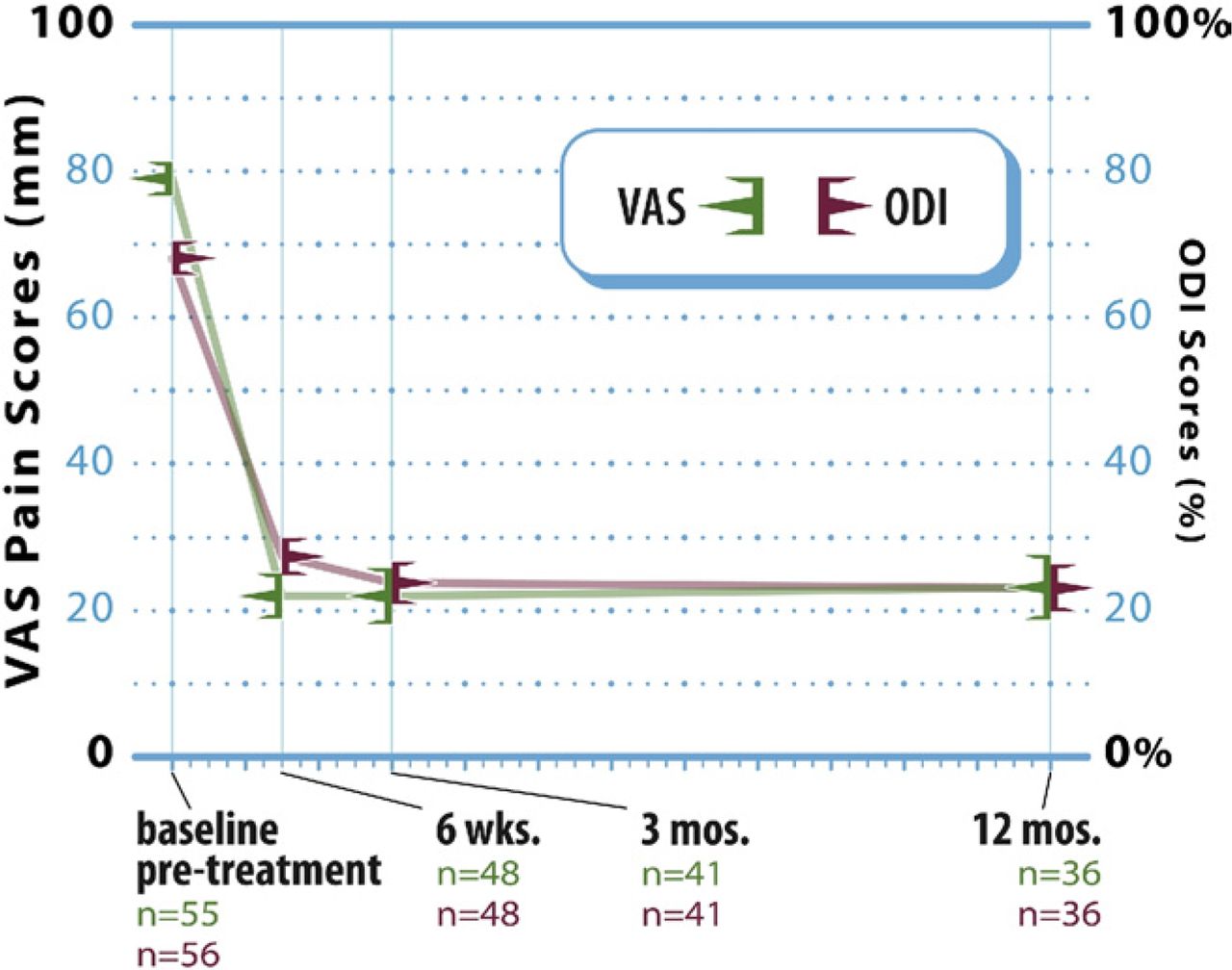
Line graph showing mean (± SE) VAS pain and ODI scores at baseline and each follow-up interval after vertebral augmentation.
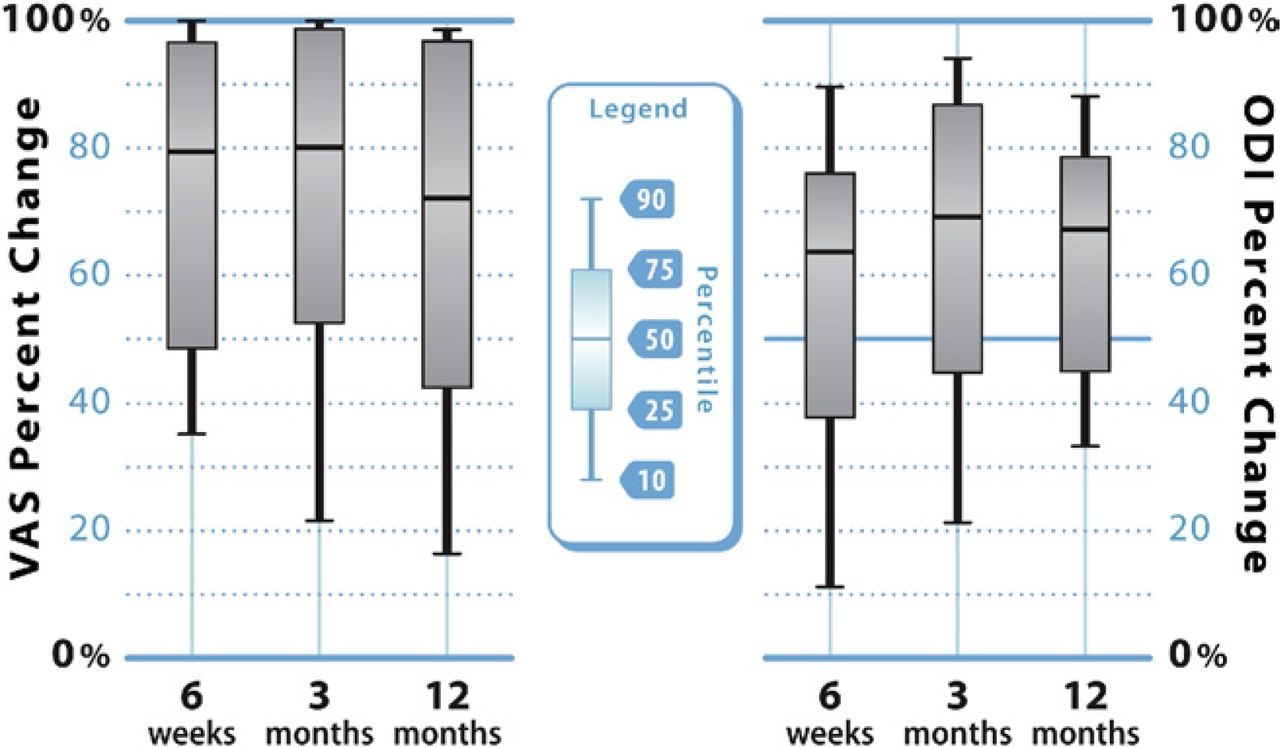
Improvement in VAS pain scores is also illustrated graphically in Fig. 4, which shows the median degree of improvement as well as the upper and lower quartiles. The median percent improvement in VAS pain scores was 79%, 80%, and 72% at 6 weeks, 3 months, and 12 months, respectively.
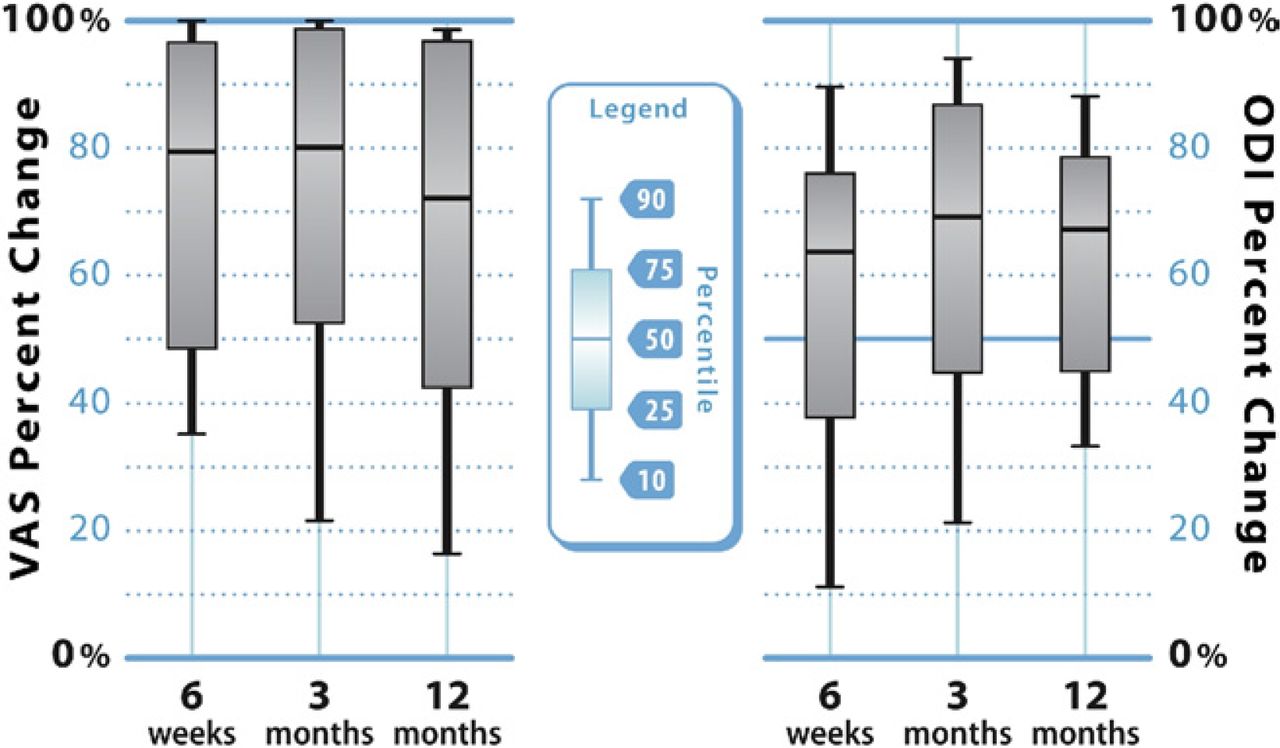
Box-and-whisker plots indicating the upper (75th) and lower (25th) quartiles and the median value for percent improvement in VAS back pain severity (left) and ODI (right) scores at 6 weeks, 3 months, and 12 months after vertebral augmentation. The points at the end of the “whiskers” represent the 90th and 10th percentile values.
Study patients showed similarly large improvements in condition-specific functional impairment after vertebral augmentation (Fig. 3). The mean ODI score improved from 68.1% ± 16.9% before treatment (n = 56) to 27.4% ± 17.2% (n = 48), 23.8% ± 18.7% (n = 41), and 23.3% ± 15.5% (n = 36) at 6 weeks, 3 months, and 12 months, respectively, which represents a mean change of 39.2 ± 19.6 percentage points, or approximately 63%, at 12 months. Again, compared with before treatment, functional status after vertebral augmentation was significantly improved at each of the 3 follow-up intervals (P < .0001 for each comparison).
Along with pain scores, improvement in ODI scores is illustrated graphically in Fig. 4. The median percent improvement in ODI scores was 64%, 69%, and 67% at 6 weeks, 3 months, and 12 months, respectively.
Overall clinical success rates were 91% (43 of 47), 88% (35 of 40), and 89% (31 of 35) at 6 weeks, 3 months, and 12 months, respectively. The vertebral augmentation procedure required injection of a mean of 2.2 ± 0.12 mL of cement per vertebral body. There were 5 of 64 levels (8%) where cement extravasation was identified radiographically, although none were related to clinical symptoms. In 30 patients (34 fractures) with adequate 12- month radiographs, 5 adjacent-level fractures, 2 nonadjacent fractures, and 1 refracture at a previously treated index level were identified.
There were no device-related adverse events reported in this study group. There was 1 procedure-related adverse event involving a dural tear that occurred during the initial pedicle access with the Jamshidi needle. A small quantity of Gelfoam was used at the site, the event resolved without incident, and there were no residual or permanent sequelae.
Discussion
There is now a large body of evidence that osteoporotic VCFs cause significant pain, functional impairment, and diminished quality of life and are harbingers of serious morbidity and an increased risk of death.12–18 Minimally invasive vertebral augmentation procedures can offer immediate and sustained symptomatic relief to patients with painful VCFs.9 It has been shown that clinical improvements in pain and function are more rapid when acutely painful VCFs are treated and stabilized percutaneously rather than managed conservatively.10
The VCF Treatment System in this study provides an innovative approach to the treatment of painful VCFs. Unlike the traditional balloon kyphoplasty procedure that pushes cancellous bone peripherally to form a repository for bone cement, the VCF Treatment System preserves cancellous architecture via a percutaneously introduced implant inserted in a continuous-loop fashion. The implant is delivered over a removable guidewire to provide structural support to the vertebral body and to serve as a conduit for bone cement placement. Vertical displacement of the endplates by the implant may result in fracture reduction. Bone cement is delivered into the preserved cancellous bone through the lumen of the implant. Excellent interdigitation as well as preferential flow of the cement toward the endplates was observed. The planar distribution of the cement supports the endplates while containment and directional control of the flow of cement minimize extravasation.
The 8% cement extravasation rate found in this study is similar to published estimates from several large literature syntheses of kyphoplasty clinical experience by Taylor et al.19 (9%), Bouza et al.20 (7%), and Eck et al.21 (7%). Strikingly, cement extravasation occurred in approximately 27% of cases in the large randomized controlled trial of Wardlaw et al.10 comparing kyphoplasty with nonsurgical care. The low extravasation rate in our study may be due in part to the relatively low volume of cement injection. In the course of treating 64 levels, a mean of 2.2 mL of cement was injected per vertebra. It appears that the volume of the implant is replacing a significant portion of cement volume that would normally be injected in a standard kyphoplasty procedure. For example, Frankel et al.22 reported a mean cement volume of 4.7 mL per vertebra. Thus the risk of extravasation may be lowered by both the containment design of the implant and the lower cement volumes used in association with the device.
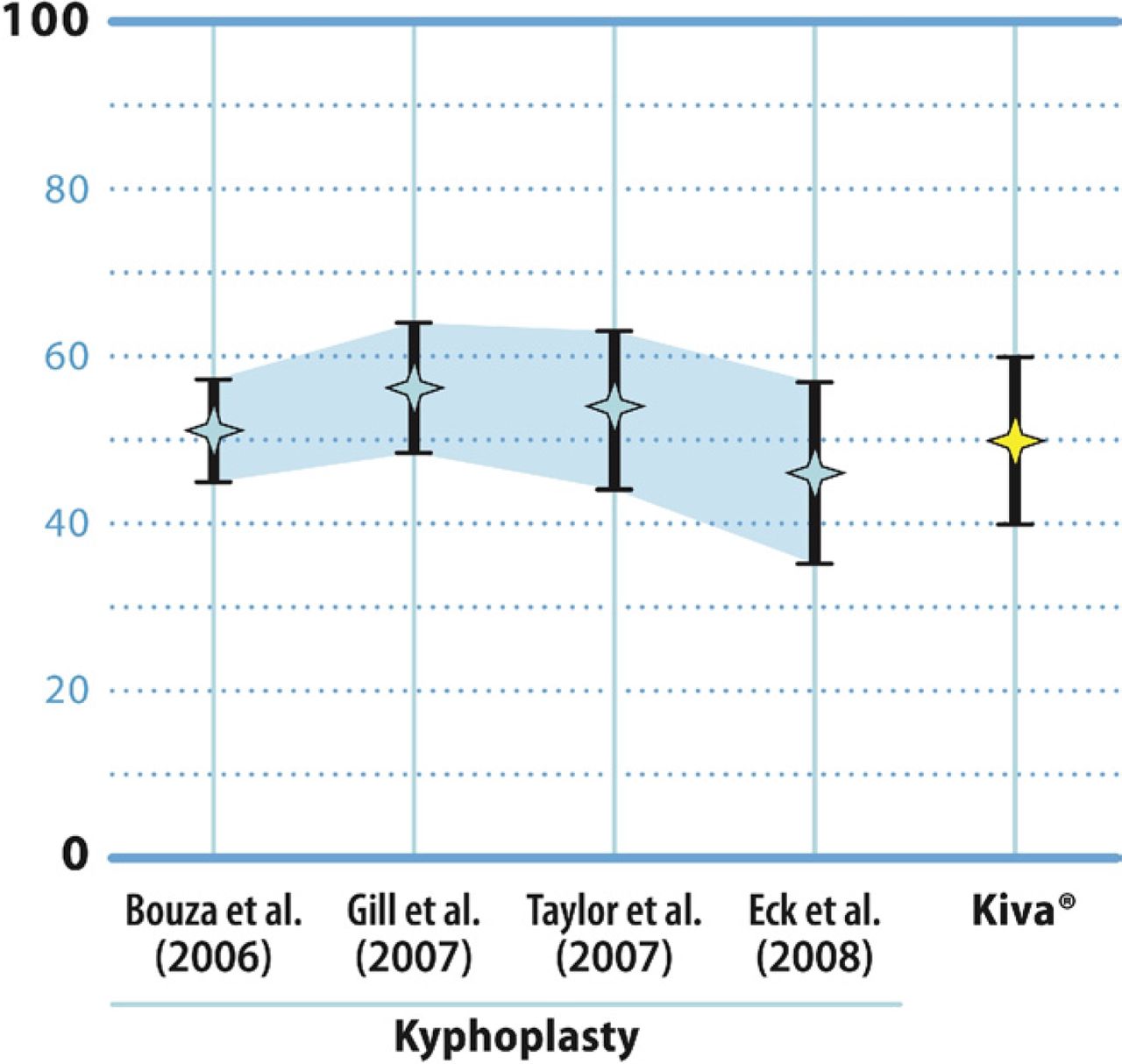
The results of this feasibility trial are encouraging. The mean reduction in postoperative pain by 12 months was approximately 50 mm on the VAS. These findings compare quite favorably with the findings from 4 separate metaanalyses of published studies of the clinical effectiveness of balloon kyphoplasty (Fig. 5). Specifically, the mean reductions reported in these meta-analyses for postoperative pain severity scores were 51 mm for Bouza et al.,20 56 mm for Gill et al.,23 54 mm for Taylor et al.,19 and 46 mm for Eck et al.21
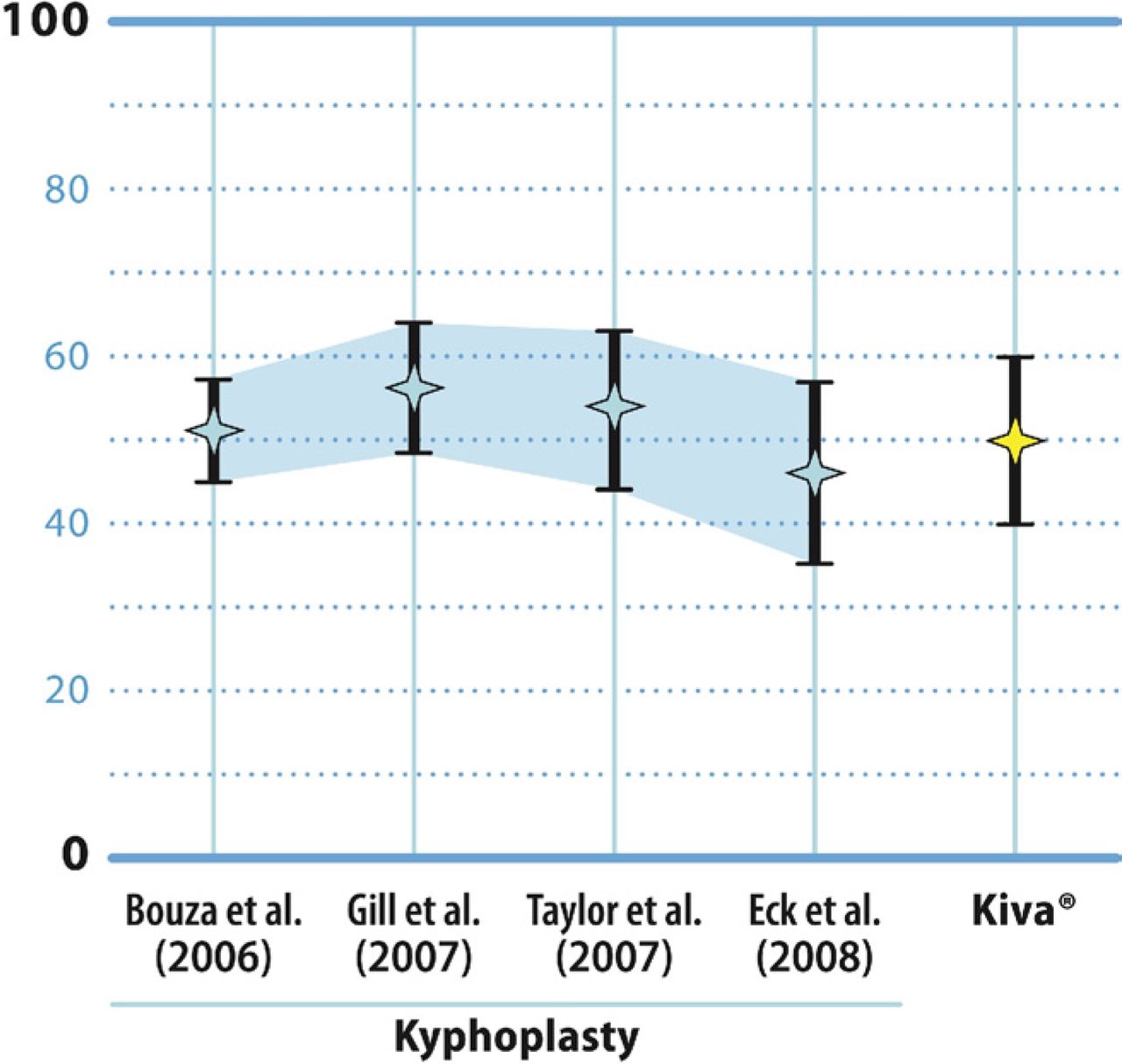
Mean and 95% confidence interval values for improvement in back pain severity by VAS derived from 4 separate meta-analyses of balloon kyphoplasty studies compared with the 12-month mean reduction in VAS pain scores for patients treated with the VCF Treatment System.
The median percent improvement in both back pain and function realized after vertebral augmentation was 64% or greater at all follow-up intervals. Thus the typical symptomatic improvement perceived by patients after this procedure was far in excess of the established minimal clinically important difference of 30% for both of these outcomes as published by Ostelo et al.11
This feasibility trial had several limitations, including the absence of an early postoperative patient follow-up interval to assess whether immediate symptomatic pain relief was achieved, as well as the lack of a concurrent control group, such as vertebroplasty or kyphoplasty, to evaluate comparative safety and effectiveness. In addition, standardized radiographic image acquisition techniques were not used, which precluded the use of automated vertebral morphometry measurements to determine the degree of height restoration.
These pilot findings show marked clinical improvement for pain and functional outcomes after this novel vertebral augmentation procedure in patients with painful VCFs. Clinically relevant gains were realized early postoperatively and maintained through 12 months of follow-up. The device could be deployed and implanted without clinically significant cement extravasation (Fig. 6).

(A) Intraoperative lateral fluoroscopic image showing moderate vertebral wedging with percutaneous initial catheter introduction. (B) Immediate postoperative image in the same patient showing complete implant deployment with cement containment.
Acknowledgments
The authors appreciate the data management support of Phil Colla and the graphical illustrations provided by Randy Asher.
- © 2011 SAS - The International Society for the Advancement of Spine Surgery. Published by Elsevier Inc. All rights reserved.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.