


Abstract
Background The Kineflex lumbar artificial disc replacement device (SpinalMotion, Mountain View, California) is a semiconstrained, posterior center of rotation, metal-on-metal intervertebral disc prosthesis. We performed a prospective, randomized, non-inferiority trial comparing the Kineflex Disc with the Food and Drug Administration (FDA)–approved Charité device (DePuy Spine, Raynham, Massachusetts). Our objective was to evaluate the Kineflex Disc's safety and efficacy using validated outcomes measures—the visual analog scale (VAS) and the Oswestry Disability Index (ODI).
Methods Sixty-four patients were randomized to receive either the Kineflex Disc or Charité device and were then followed up for up to 3 years. Patients completed VAS and ODI questionnaires and were evaluated clinically and radiologically for complication or device failure. Results were analyzed in terms of change in mean VAS score and ODI from baseline, as well as with a comparison of clinical success as defined by FDA investigational device exemption criteria. Non-inferiority was defined as a difference of less than 18 points in the VAS score and difference of less than 10 units on the ODI scale, in keeping with a previously established minimum clinically important difference.
Results The mean improvement for the Kineflex Disc group at 24 months was 56.80 for the VAS score and 37.30 for the ODI. Similarly, the mean improvement in the Charité group was 54.43 for the VAS score and 38.40 for the ODI. At 2 years of follow-up, no difference was found in VAS scores between the two groups. The Kineflex Disc group was therefore found to be non-inferior (mean difference, 2.37; 95% confidence interval, −12.5 to 17.3; P = .004). In addition, at 24 months, 83% of patients in the Kineflex Disc group and 85% of patients in the Charité group met FDA-defined criteria for clinical success, with no difference between groups (P = .802).
Conclusions This level I evidence shows the Kineflex Disc to be non-inferior to the Charité device in terms of pain reduction (VAS score) and FDA-defined clinical success at 24 months’ follow-up. Both devices showed a high degree of safety.
Lumbar artificial disc replacement (ADR) devices have been in use in the United States for several years and have generated a great deal of controversy in both the scientific literature and lay literature.1–3 Total disc replacement has previously been shown to be non-inferior to spinal fusion in terms of pain and disability reduction.4–7 Replacement, however, offers the potential advantage of maintenance of normal motion and reduced forces on adjacent vertebral levels, which may result in adjacent-level disease.8–11 Currently, 2 lumbar ADR devices are Food and Drug Administration (FDA) approved: the Charité prosthesis (DePuy Spine, Raynham, Massachusetts) and ProDisc-L prosthesis (Synthes, West Chester, Pennsylvania). Several newer devices, including the Kineflex Disc (SpinalMotion, Mountain View, California), are now seeking to gain FDA acceptance. This newer technology hopes to improve wear profiles and further decrease facet stress and shear forces while maintaining normal or near-normal range of motion.
We compared the safety and efficacy of the Kineflex Disc with the Charité artificial disc. The Charité device gained FDA approval in 2004 after the completion of several clinical trials, including a 2-year multicenter study where it was shown to be non-inferior to anterior lateral interbody fusion (ALIF) with BAK cages and iliac crest autogenous graft.12–15 Longer-term follow-up data subsequently published confirmed that non-inferiority and preservation of motion were maintained over time, and a reanalysis of the original data in 2007 suggested possible superiority throughout the 2-year follow-up.16–18
The Kineflex Disc is a 3-piece chrome-cobalt-molybdenum device, with a semiconstrained core, posterior center of rotation design, and mobile center of rotation. Its design allows 12 degrees of movement into flexion, extension, and left-/right-side bending. A retaining ring on the inferior endplate limits excursion of the captured sliding core to 2 mm in all directions and prevents dislodgment of the sliding core. The disc is introduced as a single unit with fin fixation to the vertebral endplate allowing for increased ease of implantation and improved reliability in positioning.19 A non-inferiority design was used because these features are notable only if equivalent safety and efficacy of the Kineflex Disc device can be established.
Methods
This study is a prospective, randomized, controlled noninferiority trial assessing the safety and efficacy of the Kineflex Disc to relieve symptoms associated with degenerative disc disease at either the L4–5 or L5-S1 levels. Institutional review board approval was secured before initiation of the study. Informed consent was obtained from all patients, who were then randomized to receive either a Kineflex Disc (n = 33) or Charité disc (n = 31). All patients were blinded to their device assignment until after surgery. Each was evaluated preoperatively and postoperatively, as well as at 6 weeks and 3, 6, 12, 24, and 36 months. Patients completed visual analog scale (VAS) and Oswestry Disability Index (ODI) questionnaires and were monitored clinically and radiologically for device failure, complications, and need for reoperation. Inclusion and exclusion criteria were similar to those used in the original ADR-versusfusion trials (a full review of inclusion and exclusion criteria is shown in Tables 1 and 2).
Inclusion criteria
Exclusion criteria
All data are from a single investigational device exemption center, and all surgeries were completed by 1 of 2 surgeons. Three nonrandomized Kineflex Disc training cases were performed before beginning the randomized section of the trial; however, these data were not included in the final analysis.
There were no statistically significant differences in demographic features between the 2 groups including age, sex, body mass index, smoking status, or surgical level (Table 3). The mean preoperative VAS and ODI values were 85.4 and 63.9, respectively, for the Charité group and 83.9 and 60.1, respectively, for the Kineflex Disc group, confirming that a difference in baseline pain or dysfunction was not present and that the randomization was successful. A total of 58 of 64 patients completed at least 24 months follow-up with in the specified window (+/− two months). Three patients from the Kineflex Disc group and three from the Charité group were dropped from the study or lost before this follow-up. In the Kineflex Disc group, one patient had revision to an ALIF within 24 hours of the index operation, one patient refused follow-up, and the third had an ODI that changed from 44 to 8 and a VAS score that changed from 87 to 8 at 24 months, but these were out-of-window and thus not included. In the Charité group, one patient underwent revision to a more central location of an L5-S1 implant within 24 hours of index surgery and two patients refused follow-up. One of these was seen at 5 years’ follow-up exceeding the FDA success criteria but out of window.
Study demographics
Primary endpoints
Several endpoints were chosen to evaluate a variety of criteria. FDA “clinical success” was assessed, defined as (1) a minimum of 25% improvement in low-back pain ODI score at 24 months compared with baseline; (2) no revisions, reoperations, or device removals; and (3) no occurrence of a major device-related adverse event.
We also compared mean improvement in VAS and ODI scores at 24 months. As cutoffs for non-inferiority, we used a VAS difference of greater than 18 points and an ODI score of greater than 10 units. No absolute consensus exists regarding what constitutes a minimal clinically important difference for the ODI and VAS scales. However, there is a good deal of literature suggesting that our chosen cutoffs are both theoretically sound and practical values that indicate that the patient has had significant relief (or worsening).20–24
Non-inferiority was defined as a mean reduction in VAS score in the Kineflex Disc group at 24 months of less than 18 points below the mean reduction in the Charité group. Similarly, the Kineflex Disc device would be determined to be inferior to the Charité disc if the mean reduction in the ODI score in the Kineflex Disc group was greater than 10 units below the mean improvement in the Charité group.
Secondary endpoints
Because a major benefit of disc replacement as compared with other surgical modalities is in its long-term preservation of motion and decrease in adjacent-disc disease, we performed a mixed-effects model analysis to observe the difference between the 2 groups over time. This model was used to understand the time course change over the follow-up period. The analysis accounts for the correlation between a series of observations taken on the same subject.
Surgical technique
The surgical technique for the Charité artificial disc implantation has been previously described in detail by Geisler et al.12 The Kineflex artificial disc is implanted in a similar fashion, with the following differences. The Kineflex Disc comes in 3 endplate sizes. Size 1 is 27 mm × 36.5 mm, size 2 is 30 mm × 41 mm, and size 3 is 35 mm × 44 mm. The implant is inserted as a single unit with assembled disc heights of 11 mm, 12 mm, 12.75 mm, 13 mm, and 13.75 mm. In addition, there are 3 different built-in lordosis angles of 0°, 5°, and 10°. Unique to this system are 3 distractor instruments of 9 mm, 11 mm, and 13 mm in height, as well as a disc space distractor similar to the Charité distractor and a slot cutter similar to the ProDisc-L slot-cutting technique. Although the implant is 3 separate parts, it is inserted as a single unit. The core component of the implant is a constant 10.5 mm in height with a diameter of 19.25 mm and sits within the concave surfaces of the disc endplates.
Statistical analysis
Our statistical analysis was performed in accordance with the recommendations made in the CONSORT (Consolidated Standards of Reporting Trials) consensus statement for reporting of non-inferiority trials.25 For our primary outcomes, a 2-sample t test was performed comparing the mean change in ODI and VAS scores from baseline to 2 years, as well as the percentage of each group achieving FDA-defined clinical success. Our hypothesis was that of non-inferiority; therefore the 2-sample t test was 1-sided. P values were adjusted accordingly, and the cutoff for significance ( level) was changed to .025 instead of the standard .05. Histograms were used to determine the normality of improvement scores in both the Charité and Kineflex Disc groups, for both the 2-year and 3-year analyses. Normality assumptions were adequately met for the t tests.
In the mixed-effects model analysis, the mean VAS pain score and mean ODI score were compared for the 2 groups over the 2-year follow-up. An interaction term between time and group was tested for each outcome to determine whether the difference between the groups changed over the follow-up period.
Results
In the Kineflex Disc group, the mean preoperative ODI and VAS scores were 60.09 and 83.89, respectively, whereas at 24 months postoperatively, the ODI and VAS scores were 22.79 and 27.09, respectively. For the Charité group, the ODI and VAS scores were 63.90 and 85.41, respectively, preoperatively but 25.50 and 30.98, respectively, at 24 months. One Charité patient underwent revision of her L5-S1 implant to a more central position. One Kineflex patient underwent ALIF revision of her L5-S1 implant, which had subluxated anteriorly 4 mm 24 hours after the index operation. There were no other surgical or postsurgical complications, revisions, or reoperations. Every patient was discharged within 24 hours of the operation.
Table 4 shows the mean improvement for both the VAS pain and ODI scores for the Kineflex Disc and Charité arms. The mean improvement in VAS pain score (from preoperatively to 2 years postoperatively) of the Kineflex Disc group compared with the Charité group was 2.37 (95% confidence interval [CI], −12.5 to 17.3; P = .004). Therefore it met the assumptions of non-inferiority (difference of <18 points).
Mean improvement in VAS and ODI with time
The mean improvement in ODI score from preoperatively to 24 months was −1.09 (95% CI, −12.0 to 9.9; P = .054). Because the CI is not constrained within the 10-point difference set as the criterion for significance, non-inferiority in terms of ODI is inconclusive.
At 24 months, 83% of patients in the Kineflex Disc group and 86% of patients in the Charité group met FDA criteria for clinical success. As described earlier, this is a composite endpoint that takes into account both relief of disability (as assessed by a greater than 25% reduction in ODI) and device safety (defined as no revisions, reoperations, removals, or major device-related adverse events). No statistically significant differences between the 2 groups existed in terms of clinical success (P =.802), again confirming non-inferiority.
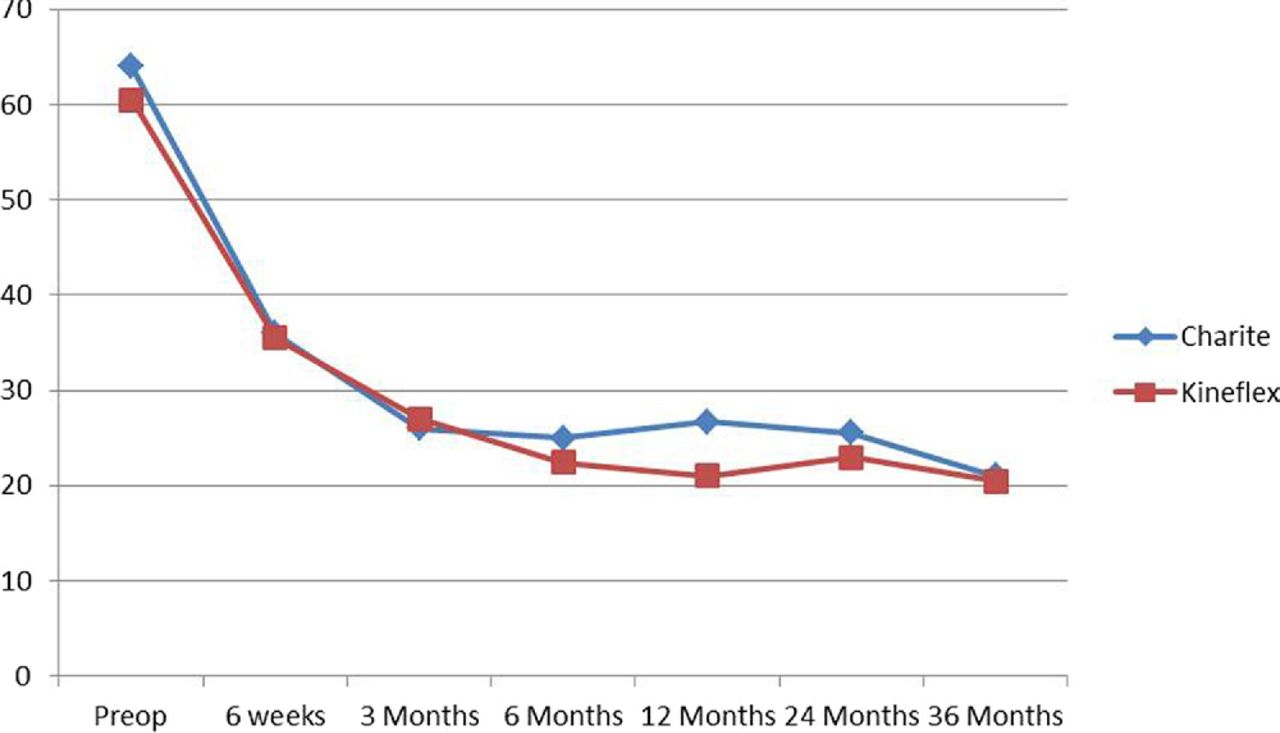
At no point in our mixed-effects model analysis was there a significant difference between the groups in terms of mean ODI or VAS score (Figs. 1 and 2). The results from this analysis show a trend toward improved pain reduction in the Kineflex Disc group. With analysis in this fashion, the Charité arm, over time, was on average 1.02 units (95% CI, −4.10 to 6.14) higher than the Kineflex Disc group (P =.69). A similar trend was noted for the disability scale, where the Charité group, over time, was on average 2.43 points (95% CI, − 2.33 to 7.20) higher than the Kineflex Disc group (P = .31).

Mean VAS pain scores over time (preoperatively, postoperatively, and at 6 weeks and 3, 6, 12, 24, and 36 months) for each group.

Mean ODI scores over time (preoperatively and at 6 weeks and 3, 6, 12, 24, and 36 months) for each group.
Discussion
The purpose of a non-inferiority trial is to use a particular intervention either as a surrogate for the gold standard or in comparing two interventions where one is believed to have an important feature that is not readily amenable to a clinical trial (price, ease of use, and so on). The Kineflex Disc was designed to be introduced as a single unit, thereby aiding reliable midline posterior introduction despite a severely diseased disc space. In addition, material studies of prostheses suggest superiority of metal-on-metal devices in terms of long-term wear profiles and of reduced immunogenicity.26–28 However, these facts are only relevant insofar as the device can be shown to be non-inferior to an established ADR device in terms of safety and efficacy.
In this study both groups showed statistical and clinically significant success over baseline for treatment of lower back pain caused by degenerative disc disease.
We found the Kineflex Disc to be non-inferior to the Charité device at 24 months of follow-up, defined as a mean reduction in VAS score by a difference of less than 18 points established by previous literature. The Kineflex Disc arm showed a small trend toward improvement over the Charité group (2.37 points); however, superiority cannot be claimed by this analysis.
The ODI analysis was inconclusive. This does not, however, indicate inferiority in terms of disability reduction. In fact, at 2 years, the ODI change CI includes a term nearing a minimum clinically significant improvement.
In addition, the mixed-effects model analysis confirms Kineflex Disc non-inferiority over time and shows a small trend toward superiority in pain and disability scores, although these conclusions cannot be definitively made.
Limitations
Our study is potentially limited by the problem of “biocreep” inherent in any multigenerational non-inferiority trial.29 With this problem, each subsequent iteration of noninferiority trial increases the size of error bars around the true results, and it becomes increasingly difficult to state with certainty that the newest intervention is, in fact, noninferior to the original (in this case ALIF with BAK cages). That said, because this procedure is no longer the standard of care, such a trial is not possible despite it being preferable in terms of statistical modeling.
In addition, a conclusion that one might be tempted to draw from this study (ie, non-inferiority of the Kineflex Disc device to spinal fusion) is limited by the fact that original trials comparing the Charité device with fusion used ALIF with BAK cages, a procedure no longer considered to be the gold standard for fusions. However, because this was the standard of care at the time of the original Charité trials, this control arm was considered reasonable.
Conclusions
The Kineflex Disc ADR device is non-inferior to the Charité ADR device at 24 months’ follow-up in terms of pain reduction and FDA-defined clinical success—a composite term that includes safety measures. The ease and reliability of implantation of the Kineflex Disc ADR device and improved wear profiles make it a viable alternative technology. Additional studies are needed to further characterize the long-term risks and benefits of these devices beyond 2-year follow-up, and a well-designed head-to-head trial is needed to establish whether any particular ADR is superior with respect to hard clinical outcomes. Every patient (except the 2 undergoing reoperations) was discharged from the hospital within 24 hours of surgery. These cases are scheduled as outpatient procedures.
- © 2011 SAS - The International Society for the Advancement of Spine Surgery. Published by Elsevier Inc. All rights reserved.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.