


Abstract
Background Interspinous process devices (IPDs) introduce a new class of complications to surgical decompression without fusion: hardware-related complications. The purpose of this study was to describe the adverse events associated with IPDs.
Study Design This was a retrospective review of the Food and Drug Administration Manufacturer and User Facility Device Experience database.
Methods The database was queried from its inception to November 2022 for reports associated with “Prosthesis, Spinous Process Spacer/Plate.” Entries were categorized by event type, patient impact, and interventions.
Results A total of 943 surgery-related adverse events were identified. The most common intraoperative events were implant malfunctions (39.7%, n = 374) and fractures (2.2%, n = 21). The most common postoperative events were persistent pain (26.6%, n = 251), implant migration (19.1%, n = 180), and fracture (6.8%, n = 64). The most common resultant outcome of an adverse event was the need for revision surgery (48.8%, n = 460). The need for revision surgery was common in patients who experienced fracture (47.1%), implant migration (84.5%), infection (76.7%), and neurological complications (76.9%). Implant migration, fracture, and implant malfunction, 3 complications that are unique to decompression with an IPD as compared with traditional laminectomy, accounted for 45.9% of revisions (211/460), and revision was required in 33.0% of cases where 1 of these complications was reported (211/640). Implant malfunction made up 21.2% of Coflex complications, 47.3% of Superion complications, and 5.2% of X-Stop complications.
Conclusions The most common adverse events were implant malfunction, inadequate efficacy, implant migration, and fracture. Concerningly, these complications require revision surgery in one-third of cases when they occur. Implant-specific assessments demonstrate a high prevalence of implant malfunctions for the Coflex and Superion implants.
Clinical Relevance Interspinous process devices introduce a new class of complications to isolated spinal decompression surgery: implant-related complications. These complications occur both intraoperatively and postoperatively, and they frequently necessitate revision surgery.
Level of Evidence 4.
Introduction
Degenerative lumbar spinal stenosis (LSS) is a common condition that affects roughly 20,000 people in the United States.1 LSS can cause neurogenic claudication, which is associated with significant impairment and diminished quality of life, making LSS the most common reason for spine surgery in the elderly population.1 Traditionally, surgical management has focused on direct central decompression at the affected level, most commonly through an open or minimally invasive (MIS) laminectomy. In an effort to minimize surgical trauma and improve recovery time, however, alternative MIS techniques have been introduced in recent years.2
One alternative MIS technique that has gained recent attention is the interspinous process device (IPD). IPDs are implants that are placed between the spinous processes at the level of pathology to relieve central stenosis.2 The reported benefits of an interspinous implant are indirect decompression of central stenosis in a manner that causes less tissue trauma and allows for shorter operative times and faster recoveries than traditional decompression techniques.3 These potential advantages, however, come with many potential pitfalls, including concerns regarding safety and efficacy.
When comparing complication rates of IPDs to traditional direct decompression techniques, such as open and MIS laminectomies, Zhao et al, Wu et al, and Welton et al have shown higher reoperation rates associated with IPDs.4–6 Multiple complications have been reported with IPDs, such as implant loosening, new or persistent pain, spinous process fractures, and implant migration, many of which require further treatment or surgical intervention.3 These complications are often caused by improper patient selection, inappropriate device size, and incorrect positioning.3,7,8 Furthermore, by introducing hardware in a procedure intended for decompression alone, IPDs introduce an entirely new class of complications to the realm of surgical decompression without fusion: implant-related complications. These implant-related complications can be so pervasive that they require a product recall, as was the case with the temporary Class II product recall associated with the X-STOP (Medtronic) device from September 2008 to July 2009.9
With the growing utilization of this technology, there is a need for continued vigilance to assess its safety and efficacy. The Food and Drug Administration (FDA) Manufacturer and User Facility Device Experience (MAUDE) database, a publicly available repository of adverse events reports, is a particularly useful tool for assessing adverse events associated with an implant. This database provides a near real-time summary of adverse events associated with new implants and technologies and allows for monitoring of their safety and efficacy. The objective of this study, therefore, was to use the MAUDE database to summarize the adverse event reports associated with interspinous implants in hopes of uncovering any concerning trends in the safety or efficacy of these devices. We hypothesized that the most common adverse event types associated with these implants would be inadequate efficacy, implant migration, and spinous process fractures.
Materials and Methods
Institutional Review Board approval was attained prior to the completion of this study (IRB 2023–1876). The FDA MAUDE database is a publicly available repository of adverse events reports voluntarily submitted by health care professionals, manufacturers, and patients, providing a comprehensive overview of adverse events associated with medical devices. The database contains voluntary adverse events reports dating from June 1993, user facility reports since 1991, distributor reports since 1993, and manufacturer reports since August 1996 for medical devices, including IPDs. Mandatory reporting to the FDA is required by manufacturers, importers, and device user facilities. Additionally, voluntary reporting is accepted by health care professionals, patients, and consumers.
We retrospectively reviewed adverse events reports in the MAUDE database regarding IPDs for the treatment of LSS. In order to gather adverse events reports, we selected “Prosthesis, Spinous Process Spacer/Plate” as the product class. All available reports from the creation of the database to November 2022 were queried and included in our analysis. For each complication, we collected the following data: type of adverse event, patient impact, and necessary interventions associated with the adverse event. Narrative reports of each adverse event were used to accurately categorize events for analysis. FDA reports relating to the Coflex (XTant Medical, Belgrade, Montana; FDA approval 10/2012), Superior (Boston Scientific, Malrborough, Massachusetts; FDA approval 5/2015), and X-Stop (Medtronic, Minneapolis, Minnesota; FDA approval 11/2005) were included in our report.
Complications were categorized into 2 groups: (1) unrelated anesthesia or medical events and (2) surgery or implant-related events. Unrelated anesthesia or medical events were eliminated from our analysis in order to focus specifically on implant-related surgical adverse events. Adverse events were further categorized as intraoperative or postoperative to allow for differentiation of events that were associated with complications during implantation vs complications that occurred as postoperative sequelae.
Adverse events were analyzed both in aggregate (across implants), as well as for individual implant manufacturers (within implants). This allowed us to determine what proportion of each adverse event type was attributable to each implant, as well as to determine the relative representation of each adverse event for a given implant. No tests for statistical significance were performed, but counts of adverse events were summed, and proportions were calculated using SPSS statistical software (IBM Corp., Armonk, NY).
Results
Overall Results
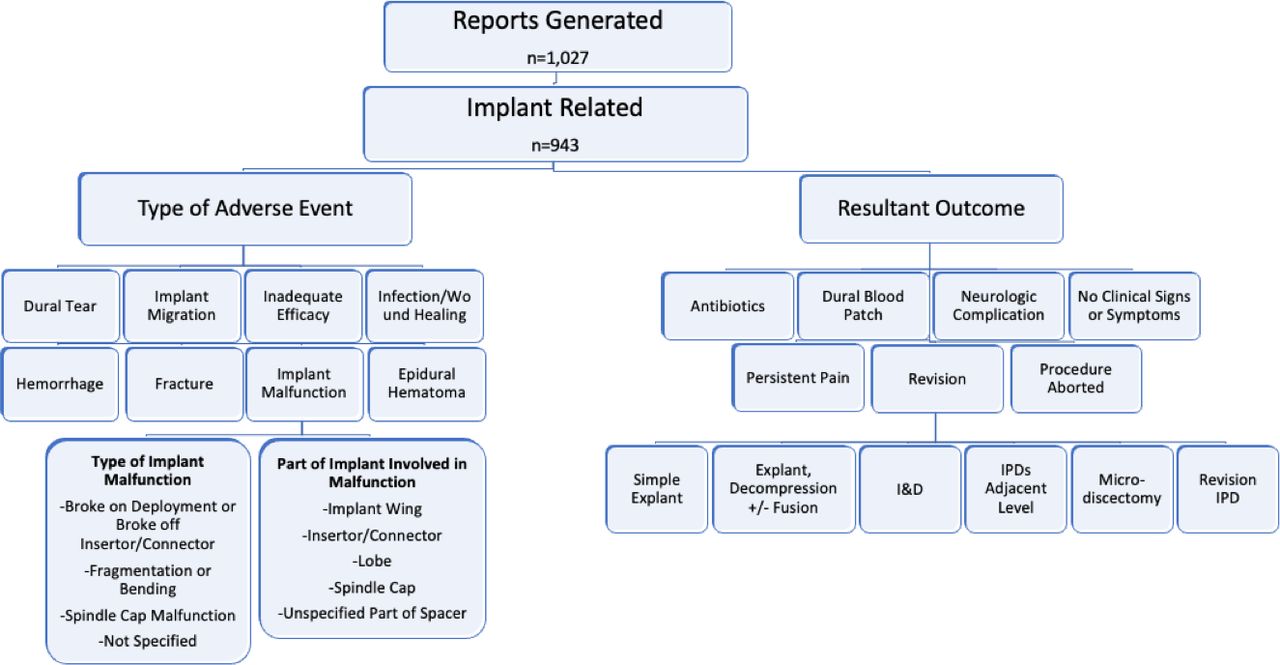
Our search yielded a total of 1027 adverse events reports. Thirty-nine of these reports were generated from published abstracts or manuscripts and were eliminated due to the high likelihood that these could represent duplications of previously submitted reports. Five were technical errors (wrong-level surgery) and were also eliminated. This yielded 983 reports for analysis. Forty of these events were unrelated anesthesia or medical events, while the remaining 943 were surgery related (Figure 1).

Summary of reports generated from the Manufacturer and User Facility Device Experience database for “Prosthesis, Spinous Process Spacer/Plate.”
Of the 943 surgery-related adverse events, 42.8% (404) were detected intraoperatively and 57.2% (539) were postoperative sequelae. The most common intraoperative events were implant malfunctions (39.7%, n = 374) and fractures (2.2%, n = 21). The most common postoperative events were persistent pain (26.6%, n = 251), implant migration (19.1%, n = 180), and fracture (6.8%, n = 64; Table 1). The 2 most common types of implant malfunctions, which were unique to the Superion implant, were spindle cap malfunction during deployment (52.1%, n = 195) and implant breakage off of the insertor/connector (35.8%, n = 134; Table 2).
Intraoperative and postoperative adverse events related to interspinous process devices.
Malfunctions of interspinous process devices.
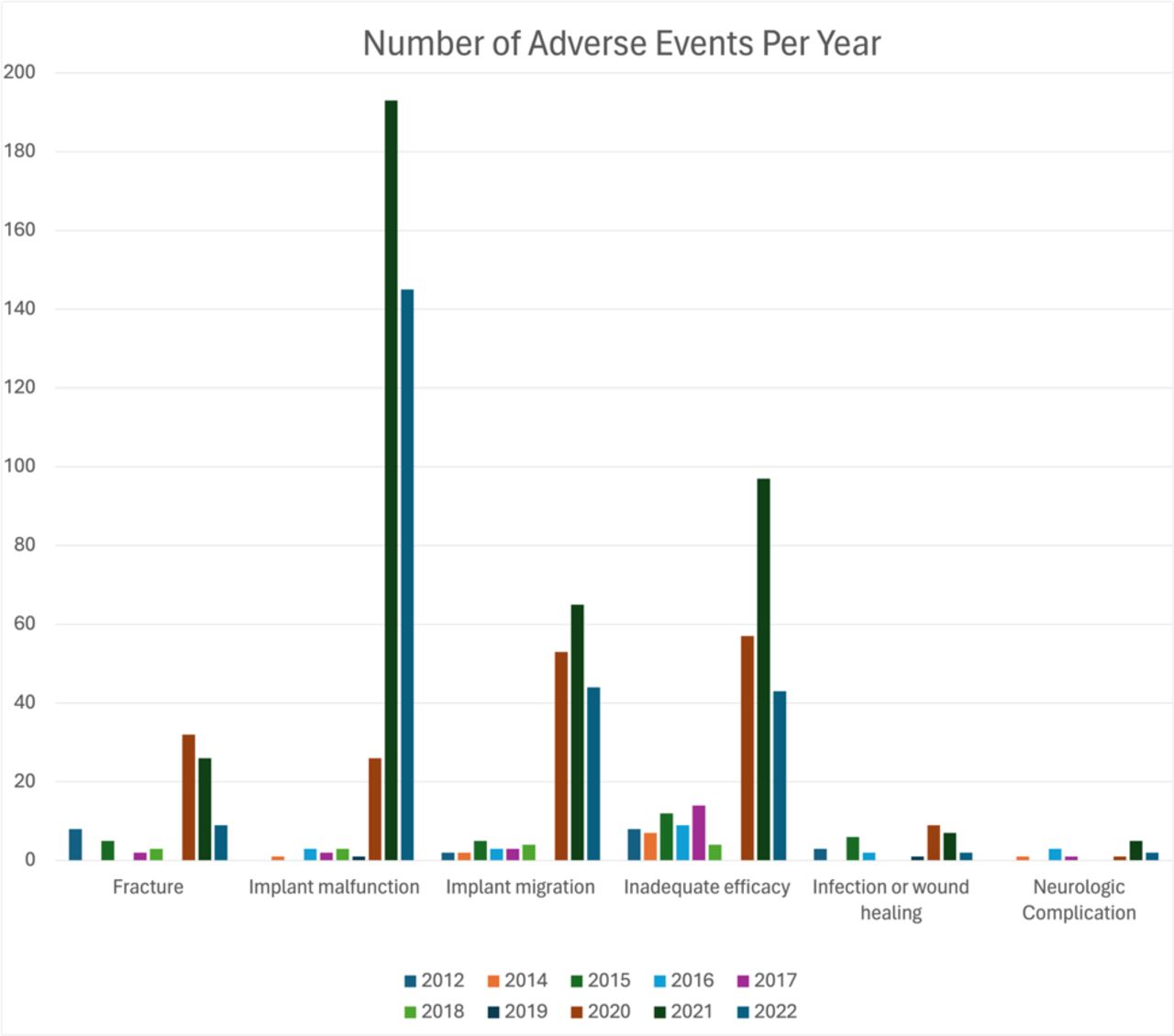
The resultant outcome of each adverse event is summarized in Table 3. While a large number of adverse events had no clinical impact on the patient (aside from unmeasured impacts, such as increased operative time and cost; 34.8%, n = 328), the most common resultant outcome of an adverse event was the need for revision surgery (48.8%, n = 460). The following most common outcomes were persistent pain (13.0%, n = 123) and the need to abort the index procedure (34.8%, n = 328). The need for revision surgery was common in patients who experienced fracture (47.1%), implant migration (84.5%), infection (76.7%), and neurological complications (76.9%; Table 3). Trends in resultant outcomes over time are presented graphically in Figure 2 and demonstrate increasing numbers of adverse events, especially for implant malfunction, implant migration, and inadequate efficacy.

Number of adverse events of each type per year.
Resultant outcome as a function of adverse event type related to interspinous process devices.
Among the patients who required revision surgery, inadequate efficacy (46.7%, n = 215), implant migration (33.3%, n = 153), and fracture (8.7%, n = 40) were the most common indications (Table 4). Implant migration, fracture, and implant malfunction, 3 complications that are unique to decompression with an IPD as compared with traditional laminectomy, accounted for 45.9% of revisions (211/460), and revision was required in 33.0% of cases where 1 of these complications was reported (211/640).
Reasons for revision after placement of interspinous process devices.
Among patients who had the index procedure aborted, fracture (61.1%, n = 11), implant malfunction (16.7%, n = 3), and dural tears (11.1%, n = 2) were the most common reasons. The most common revision procedures were simple explant (59.9%, n = 226), revision IPD (22.3%, n = 84), and explant with decompression with or without fusion (14.3%, n = 54; Table 5).
Type of revision after interspinous process device placement.
Implant-Specific Events
Coflex
There were 38 adverse events reports associated with the Coflex device, accounting for 4.0% of all adverse events reports. A total of 21.1% were intraoperative, while 78.9% were postoperative (Table 1). The most common complications were inadequate efficacy (28.9%, n = 11), implant malfunction (21.1%, n = 8), and infection or wound healing complications (21.1%, n = 8; Table 3). There were 8 reports of implant malfunction, all of which were due to fragmentation/bending (Table 2).
The most common resultant outcomes were revision (81.6%, n = 31) and persistent pain (15.8%, n = 6). The need for revision surgery was common in patients who experienced implant malfunction (87.5%), fracture (80.0%), implant migration (60.0%), infection (87.5%), and neurological complications (100.0%; Table 3). Implant migration, fracture, and implant malfunction accounted for 45.2% of Coflex revisions (Table 4). Among the patients who required revision, the most common revision types were simple explant (40%, n = 12), explant with decompression with or without fusion (30%, n = 9), and revision interspinous implants (20%, n = 6; Table 5).
Superion
There were 828 adverse events reports associated with the Superion device, accounting for 87.8% of all adverse events reports. A total of 47.3% were intraoperative, while 52.7% were postoperative (Table 1). The most common complications were implant malfunction (44.1%, n = 365), inadequate efficacy (23.9%, n = 198), and implant migration (19.6%, n = 162; Table 3). Of the 365 reports of implant malfunction, the most common were spindle cap malfunction during deployment (53.4%, n = 195), implant breaking on deployment or breaking off of the connector/insertor (36.7%, n = 134), and implant fragmentation/bending (6.8%, n = 25; Table 2). The most common parts of the implant involved in malfunction were the spindle cap (53.7%, n = 196), an unspecified part of the spacer (40.0%, n = 146), and the insertor/connector (4.9%, n = 18).
The most common resultant outcomes were revision (46.3%, n = 383), no clinical signs or symptoms (39.1%, n = 324), and persistent pain (11.0%, n = 91). The need for revision surgery was common in patients who experienced implant migration (86.4%), fracture (39.7%), infection (66.7%), and neurological complications (62.5%; Table 3). Implant migration, fracture, and implant malfunction accounted for 46.2% of Superion revisions. Unlike with the Coflex, the risk of revision associated with implant malfunction was low (2.6%), but migration, fracture, and implant malfunction accounted for 46.2% of Superion revisions (Table 4). Among the patients who required revision, the most common revision types were simple explant (67.1%, n = 200), revision interspinous implants (24.8%, n = 74), and explant with decompression with or without fusion (5.7%, n = 17; Table 5).
X-Stop
There were 77 adverse events reports associated with the X-Stop device, accounting for 8.2% of all adverse events reports. A total of 5.2% were intraoperative, while 94.8% were postoperative (Table 1). The most common complications were inadequate efficacy (54.5%, n = 42), implant migration (18.2%, n = 14), and fracture (15.6%, n = 12; Table 3). There was 1 report of implant malfunction, the cause of which was not specified.
The most common resultant outcomes were revision (59.7%, n = 46), persistent pain (33.8%, n = 26), and no clinical signs or symptoms (5.2%, n = 4). The need for revision surgery was common in patients who experienced fracture (75.0%), implant malfunction (100.0%), implant migration (71.4%), infection (100.0%), and neurological complications (100.0%; Table 3). Unlike with the Coflex, the risk of revision associated with implant malfunction was low (2.2%), but migration, fracture, and implant malfunction accounted for 43.5% of X-Stop revisions (Table 4). Among the patients who required revision, the most common revision types were explant with decompression ± fusion (57.1%, n = 54), explant (28.6%, n = 14), and revision interspinous implants (8.2%, n = 4; Table 5).
Discussion
IPDs have emerged as a popular treatment option for patients with degenerative LSS, providing a less invasive alternative to traditional surgical interventions.2 However, the safety and efficacy of these implants have been a topic of debate within the orthopedic community. In the current study, we assessed 943 adverse events that were reported to the FDA and detailed in the MAUDE database. Implant malfunctions (39.7%), inadequate efficacy (26.6%), implant migration (19.2%), and fracture (9.0%%) were the most common postoperative adverse events. Among the most common adverse events, revision surgery was often required for definitive treatment (fracture 47.1%, implant migration 84.5%, and inadequate efficacy 85.7%).
There were implant-specific differences in the representation of various adverse events. While fracture, implant migration, and inadequate efficacy represented a large proportion of the adverse events in each group, there were large differences in the proportion of certain adverse events. Implant malfunction, for example, represented a large proportion of the adverse events for the Superion (44.1%) and Coflex (21.1%) but only a small proportion for the X-Stop (1.3%). The high proportion of implant-related malfunctions in the Superion group may reflect a systematic issue with the intraoperative placement or deployment of these devices. Given the high proportion of reports indicating malfunctions related to the spindle cap and the spacer implant itself, it is likely that 1 or both of these components have mechanical or technical shortcomings that have contributed to the high proportion of mechanical failures for this implant. Interestingly, while the X-Stop implant underwent a temporary Class II recall in 20099 due to “potential to cause damage to and/or breakage of the…device’s universal wing assembly,” there have been no such recalls for the Superion despite the high number of the implant malfunctions identified in the current study. While implant malfunctions only required revision in 2.7% of the Superion cases, they required revision in 87.5% of Coflex reports.
Another implant-specific difference is that a far higher proportion of the adverse events reports for the X-Stop relate to inadequate efficacy (54.5%) in comparison to the Coflex (28.9%) and Superion (23.9%). Such disparities may highlight important shortcomings related to the degree of relief provided by these implants.
One of the most important findings in the current study is the high likelihood of revision surgery for complications that specifically can be attributed to the IPD. Because spinous process fractures, implant migration, and implant malfunction are implant specific, these represent a fairly unique adverse event class compared with traditional uninstrumented laminectomy for decompression. Implant migration, implant malfunction, and fracture accounted for roughly 45% of revisions, and one-third of cases where these adverse events were present required a revision surgery.
The results of the current study corroborate the findings of multiple clinical trials reporting adverse events associated with IPDs. In a 2-year study assessing clinical outcomes of the X-Stop device, Hartjen et al10 identified a 3.6% risk of persistent pain, 1.8% risk of device migration, and a 1.8% risk of spinous process fracture. Nearly 10% of these patients required explant with or without further decompression within the study period due to adverse events or inadequate efficacy. Similarly, Verhoof et al11 found a 58% revision rate within 2 years following insertion of the X-Stop spacer. In a clinical study assessing the Superion device, a total of 44.4% of patients experienced device-related complications, including device migration (11.1%) and spinous process fractures (21.1%). A total of 20.1% of patients required device explantation, and 24.3% required same-site revision surgery.6 Taken together, the results of this analysis and prior studies demonstrate a concerning number of adverse events associated specifically with the efficacy and safety of IPDs that should lead treating physicians to question the use of these devices over standard uninstrumented decompression techniques.
We believe that while the current study cannot make definitive conclusions regarding the safety and efficacy of these implants, it does alert surgeons to the potential adverse events associated with IPDs. This is especially useful in the setting of implant-related complications, which were common. Surgeons should be aware that implants often malfunction on deployment or cause spinous process fractures on deployment. Specific technical pearls can also be derived from these data. For example, surgeons should be aware that Coflex implants are subject to intraoperative fragmentation and bending, while Superion implants are more prone to spine cap malfunction, implant breakage on deployment, and implant breakage off the insertor/connector. These findings may help surgeons determine whether they would like to perform IPDs, which implants they might prefer, and which technical aspects of the surgical procedure require additional caution or vigilance.
This study had many limitations. First, because the MAUDE database lacks data on the number of implants placed over the period under analysis, it is difficult to ascertain a prevalence or incidence. Therefore, it is difficult to compare the number of adverse events reports between implants in any meaningful way, as a high number of adverse events reports may simply be a reflection of a higher utilization of a given implant. This makes it impossible to comment directly on the comparative safety and efficacy of these implants. Second, the MAUDE database relies on voluntary reporting, which may lead to underrepresentation of certain adverse events. Third, this database lacks patient- and surgery-specific details that may contribute to the occurrence of an adverse event, such as bone quality, anatomical abnormalities, or other conditions. Finally, because these implants are often placed by nonsurgeon physicians, it is possible that technical errors surrounding proper implant placement techniques may be partially responsible for many of the adverse events seen in this report, including implant malfunction, fractures, and implant migration.
Conclusions
Per review of the FDA MAUDE database for adverse events, there are a large number of adverse events reports associated with IPDs. The most common reports are for implant malfunction, inadequate efficacy, implant migration, and fracture. While inadequate efficacy and continued pain are possible with traditional decompression techniques, the use of IPDs introduces hardware-related complications to the realm of surgical decompression without fusion (implant malfunction, implant migration, and fracture). Concerningly, these complications require revision surgery in one-third of cases. Furthermore, implant-specific assessments demonstrate a high prevalence of implant malfunctions for the Coflex and Superion implants. While these rarely led to revision surgery in the Superion, nearly 90% of Coflex implant malfunctions led to revision surgery.
Footnotes
Funding The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests The authors report no conflicts of interest in this work.
Disclosures Dr. Qureshi reports relationships with AMOM opportunities (honoraria), Globus Medical, Inc. (speakers' bureau, consultant, and royalties), HS2, LLC (ownership interest), HSS ASC Development Network, LLC (ownership interest), Stryker K2M (consultant, royalties and designer), Tissue Differentiation Intelligence (ownership interest), See All AI (ownership interest, scientific advisory board), and Viseon, Inc. (research support, consultant). Dr. Iyer reports relationships with HS2,LLC (ownership interest and editorial board), HSS ASC Development network, LLC (ownership interest), Joint Effort Administrative Services Organization, LLC (ownership interest), Innovasis (consultant), and Intrinsic Therapeutics (consultant). The remaining authors have no disclosures.
- This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery. Copyright © 2024 ISASS. To see more or order reprints or permissions, see http://ijssurgery.com.
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.