


Abstract
Background Anterior cervical fusion, an established procedure to treat cervical radiculopathy, sacrifices the natural function of the disc, while placing increased stresses on adjacent spinal levels. In contrast, the cervical total disc replacement (cTDR) maintains motion and decreases adjacent-level stresses. The purpose of this study was to investigate the safety and effectiveness of a next-generation cTDR device in patients with symptomatic cervical radiculopathy.
Methods This is a multicenter Food and Drug Administration–regulated feasibility study to evaluate safety and effectiveness of the M6-C Artificial Cervical Disc for the treatment of patients with symptomatic cervical radiculopathy at 1 or 2 levels from C3 to C7. Neck Disability Index (NDI), visual analog scales (VAS) assessing neck and arm pain, Short Form 36 Health Survey (SF-36), safety, and radiographic outcomes were assessed preoperatively, at 6 weeks and 3, 6, 12, and 24 months postoperatively.
Results Thirty patients were enrolled at 3 clinical sites. Patients were implanted at either 1 or 2 levels. Mean NDI improved from 67.8 to 20.8 (P < .0001) at 24 months. Significant improvement was also observed through 24-month follow-up in neck and arm pain VAS (P < .0001) and in physical (P < .005) and mental component scores of the SF-36 at 3, 6, and 12 months (P < .008). There were no serious adverse events related to the device or procedure as adjudicated by an independent clinical events committee. Radiographically, disc space height increased more than 50% with a correlative increase in the postoperative disc angle. Range of motion decreased slightly from baseline during early follow-up but increased slightly and were maintained throughout the follow-up period.
Conclusions The M6-C cervical artificial disc represents a new generation of cTDR design. Results of this study found the M6-C device to produce positive clinical and radiographic outcomes similar to other cTDRs, warranting further investigation.
Primary symptoms of cervical radiculopathy are neck and arm pain, as well as neurologic symptoms, including motor, reflex, and sensory changes. Anterior cervical fusion (ACF) is a well-accepted surgical procedure to treat acute cervical radiculopathy resistant to nonsurgical treatment. However, a paradigm change from fusion to motion preservation is occurring in the treatment of a subset of patients with symptomatic cervical radiculopathy. The practical advantages of cervical total disc replacement (cTDR) include maintenance of intervertebral motion and decreased adjacent-level stresses, with the theoretic advantage of reduction of adjacent-level clinical and radiographic degeneration.1–5 It has been estimated that as many as 90% of fusion-treated patients will have new-onset or progressive degenerative changes at adjacent levels, with approximately 25% of these patients exhibiting symptoms within 10 years.6 The incidence of symptomatic adjacent-level disease after ACF requiring further intervention is generally quoted as 2% to 3% annually.6–8
The clinical outcomes of previously published prospective, randomized cTDR studies have been positive.4, 9–14 In these studies comparing total disc replacement (TDR) with ACF, the results have been consistent and have found that both groups improved significantly compared with their preoperative condition, with cTDR equivalent or superior to ACF in overall success rates.15 To date, all of these devices have produced motion through a ball-and-socket–type design, with some incorporating a sliding mobile core into the design. Next-generation cTDR devices have been designed to produce and preserve motion through compression of the prosthetic core. The purpose of this study was to investigate the safety and therapeutic effectiveness of 1 such device in a US Food and Drug Administration (FDA)–regulated pilot study in patients with symptomatic cervical radiculopathy with 24-month follow-up.
Methods
Device description and surgical technique
The M6-C Artificial Cervical Disc (Spinal Kinetics, Sunnyvale, California) is a novel, next-generation artificial disc designed to replicate the anatomic, physiological, and biomechanical characteristics of the native disc. The core is composed of a polycarbonate urethane polymeric material that is surrounded by a polyethylene woven-fiber construct. The compressible polymer core is designed to simulate the stiffness and function of the nucleus, and the fibers are designed to simulate the annulus. The fiber annulus is an assembly of high–tensile strength, ultrahigh-molecular-weight polyethylene fibers wound in multiple redundant layers around the polymer nucleus, providing progressive resistance to motion. The core construct is attached to titanium alloy endplates to form the cervical disc prosthesis. This design enables the device to have all 6 df, including axial compression with independent angular motions in flexion-extension, lateral bending, and axial rotation, allowing independent translations along the 3 anatomic axes.
The prosthetic disc also has a polymer sheath encasing the core and fiber construct to inhibit any tissue ingrowth as well as capture any potential wear debris. The endplates are attached to the vertebral body by 3 low-profile keels on the superior and inferior surfaces. The endplates and keels are coated with porous titanium to promote bonecontact surface area and osseointegration (Fig. 1).

Cutaway view of single-piece M6-C compressible prosthesis.
The surgical implantation of the disc consists of a straightforward 3-step process: (1) trialing, where a foot-print template and trial implant are used to determine the appropriate size and position of the implant as well as for midline verification; (2) keel cutting, where a chisel is used to create keel tracks into the superior and inferior vertebral bodies; and (3) implant insertion, where an implant inserter is used to place the artificial disc into the desired position within the intervertebral space. Access to the disc space is accomplished through a standard anterior approach with anterior discectomy and decompression being performed. Fluoroscopic guidance is used during artificial disc placement.
Study Rationale and Design
This is an FDA-regulated Investigational Device Exemption feasibility study conditionally approved for initiation in the United States in December 2007 with final approval received on March 12, 2008 (G050254/S003). The primary study objective was to evaluate the initial safety and effectiveness of the M6-C artificial cervical disc in the treatment of patients with symptomatic cervical radiculopathy at 1 or 2 levels from C3 to C7 requiring surgery. The study was a multicenter, prospective single-arm investigation. Thirty patients were enrolled at 3 clinical sites. Data were collected preoperatively, in the operating room, and at 6 weeks and 3, 6, 12, and 24 months postoperatively.
Inclusion/exclusion criteria
All patients were skeletally mature (21–75 years old) with intractable cervical radiculopathy with or without spinal cord compression (Nurick classification ≤2) at 1 or 2 levels from C3 to C7. Patients had cervical root compression visualized on magnetic resonance imaging; concordant symptoms and signs of cervical root–related dysfunction, pain, or both; and persistent pain despite nonoperative management for at least 6 weeks.
Patient population
Patients were enrolled in the study according to specific inclusion/exclusion criteria typical for patients presenting with symptomatic cervical radiculopathy. All patients signed a study-specific institutional review board–approved informed consent form before participation. Thirty patients entered the study, and 28 (93%) completed the 24-month follow-up. One patient, incarcerated sometime after the 3-month visit, was removed from the study as recommended by the reviewing institutional review board. One patient chose to discontinue participation before the 24-month visit.
Clinical outcomes
Clinical effectiveness measures included the Neck Disability Index (NDI), neck and arm pain visual analog scales (VASs), and Short Form 36 (SF-36) Health Survey. The NDI measures neck pain, function, and disability; scores were calculated as a percentage ranging from 0% to 100%.16
Neck pain intensity and arm pain intensity were evaluated separately using VASs. The VAS is a 10-cm horizontal line anchored on the left by the words “no pain” and on the right by the words “worst possible pain.” Patients were presented with 3 VASs, 1 each for neck pain, right arm pain, and left arm pain. Patients were asked to place a mark on the line at the point that indicated their current pain intensity. The SF-36 Health Survey is a validated quality-of-life assessment.17 The physical and mental component scores (physical component summary [PCS] and mental component summary [MCS]) were calculated.
Safety assessment
Patients were monitored for the occurrence of adverse events intraoperatively and throughout follow-up. All adverse events, including implant and/or fiber breakage, sheath dislodgement, subsidence, migration, expulsion, and additional surgical procedures, including supplemental fixation, revision, and device removal, were documented. Serious adverse events (SAEs) related to the implantation procedure or to the device were captured on a continuous basis and reviewed and adjudicated by an independent clinical events committee composed of 3 spine surgeons who were not participating in the study as investigators.
Subject overall success
Overall subject study success was defined as (1) no device or implantation procedure–related SAEs; (2) no additional surgical intervention at the operative level, including supplemental fixation, revision, and/or device removal; and (3) minimum 15-point improvement in NDI scores.
Radiographic outcomes
Standard radiographs (anteroposterior, lateral, flexion, extension, and left and right lateral bending) were taken preoperatively and at all scheduled follow-up visits. A core radiographic laboratory (Medical Metrics, Inc., Houston, TX) performed a radiographic assessment according to a radiographic evaluation protocol. Primary radiographic outcomes include disc height, disc angle, index-level range of motion (ROM), global ROM (C2-6), and degree of lateral bending.
Statistical methods
Significance values for change from baseline for continuous measures such as VAS scores and NDI were based on the paired t test, and between-group (1 vs 2 levels treated) comparisons for such measures were based on the 2-sample t test. Between-group comparison of overall success was performed with the Fisher exact test. All significance values presented are 2 tailed. All statistical results were obtained using SAS, version 9.1 (SAS Institute Inc., Cary, NC). A normal distribution was assumed for the radiographic outcomes, and analysis of variance with post hoc Bonferroni comparisons was conducted between subgroups.
Results
There were 19 men and 11 women enrolled in the study. The mean age was 45.1 years, and the mean symptom duration before surgery was 2.4 years. The mean body mass index was 28.3 kg/m2. As indicated previously, patients were implanted at either 1 (n = 12) or 2 (n = 18) levels from C3 to C7 depending on their presenting condition. The majority of implants involved the C5-6 level (56.2%), with the remainder at C6-7 (29.2%) or C4-5 (14.6%).
Surgery outcomes
Estimated blood loss, surgery duration, and length of hospital stay for 1- and 2-level cases are shown in Table 1.
Perioperative data for 1- and 2-level disc replacements
Clinical outcomes
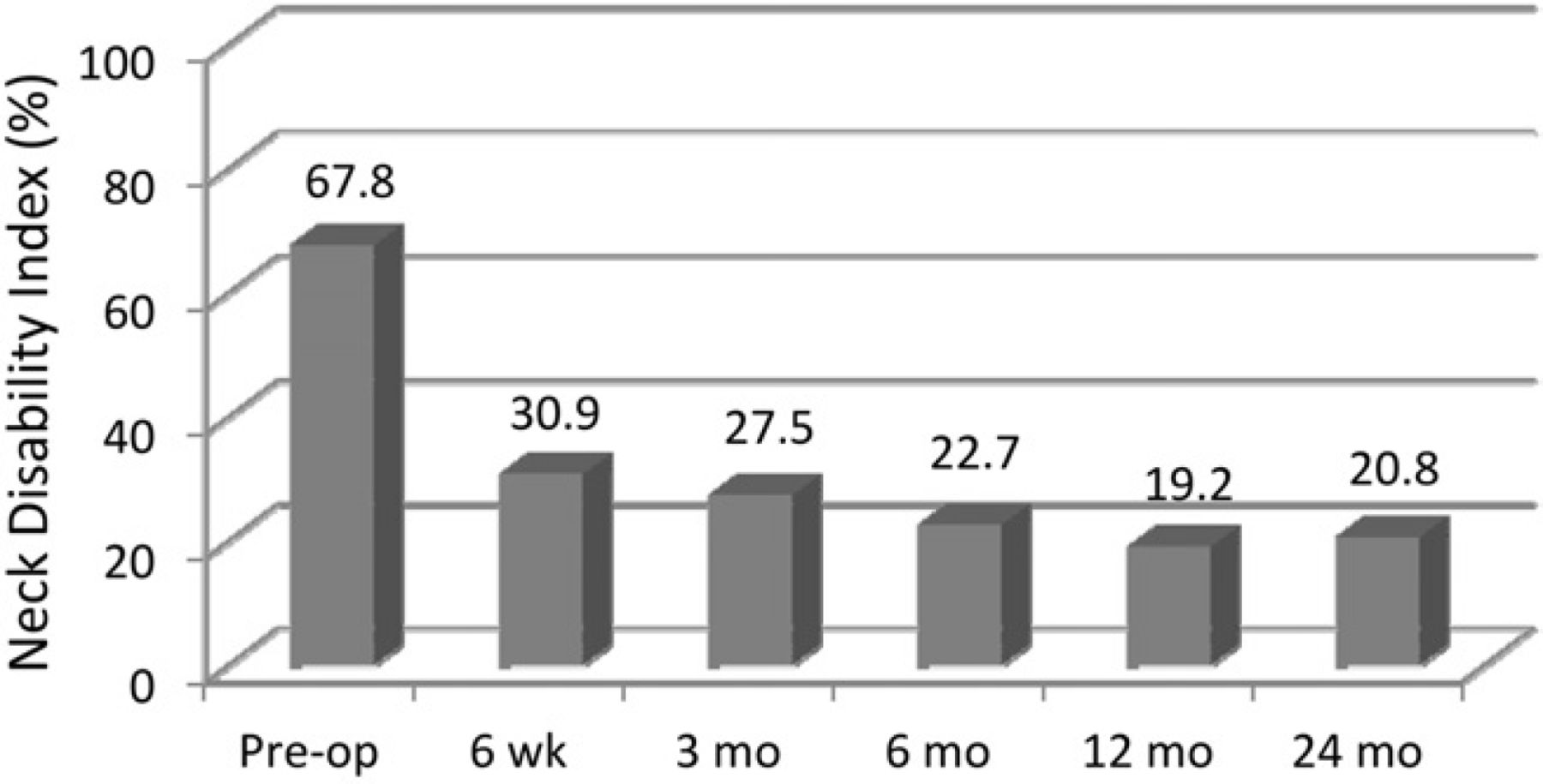
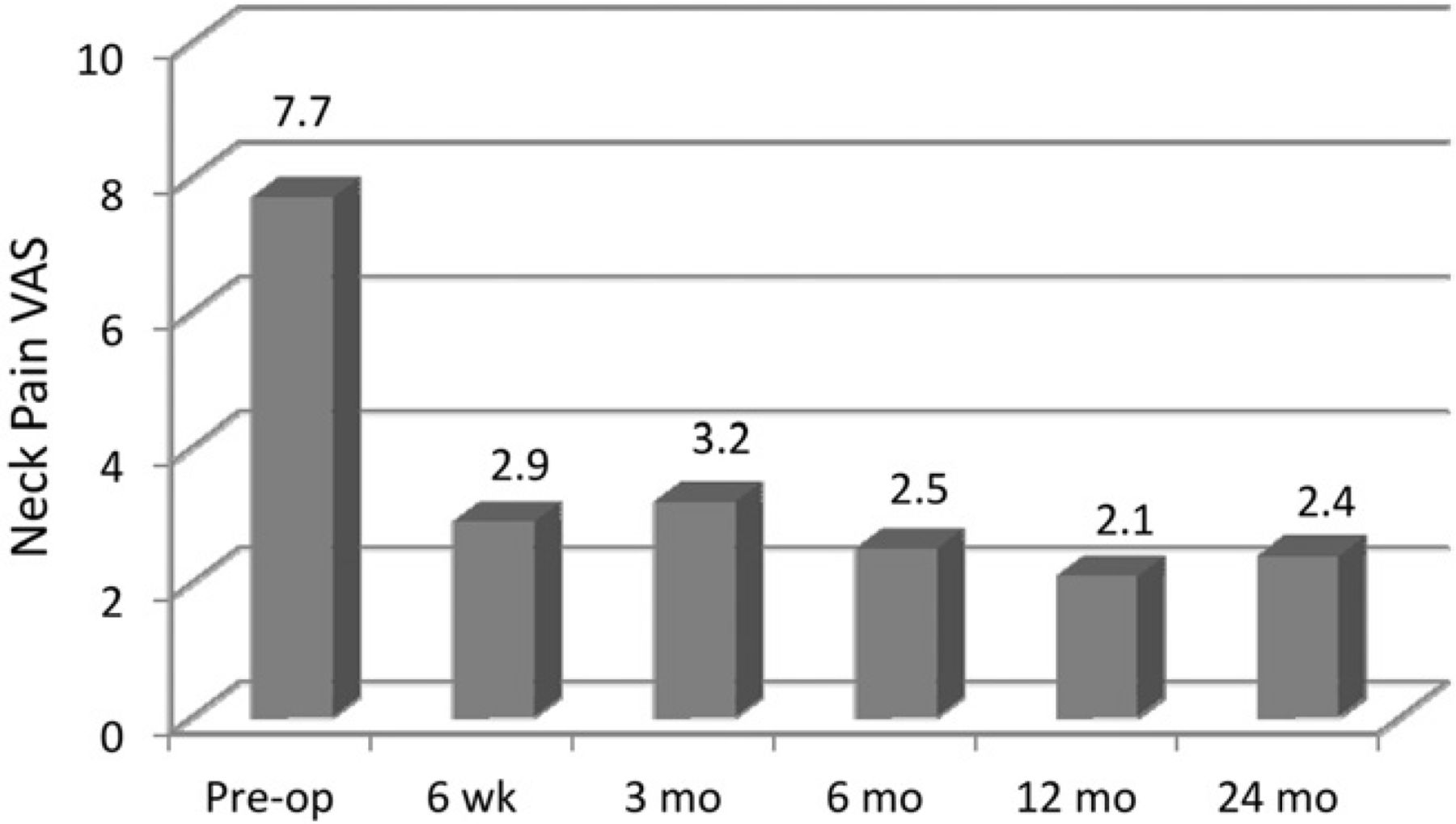
Compared with the mean preoperative value, the mean NDI scores were significantly improved by 6 weeks’ follow-up and remained so throughout the 24-month follow-up (P < .001) (Fig. 2). Figures 3 and 4 show the improvement in the mean preoperative VAS scores compared with the postoperative mean scores for both neck and arm pain. The improvements in neck and arm pain VAS were significant (P < .01) throughout follow-up. The PCS and MCS scores on the SF-36 were significantly improved from baseline at all follow-up evaluations with the exception of the MCS at 24-month follow-up (Table 2).

Mean NDI through 24-month follow-up. (Pre-op, preoperatively.)
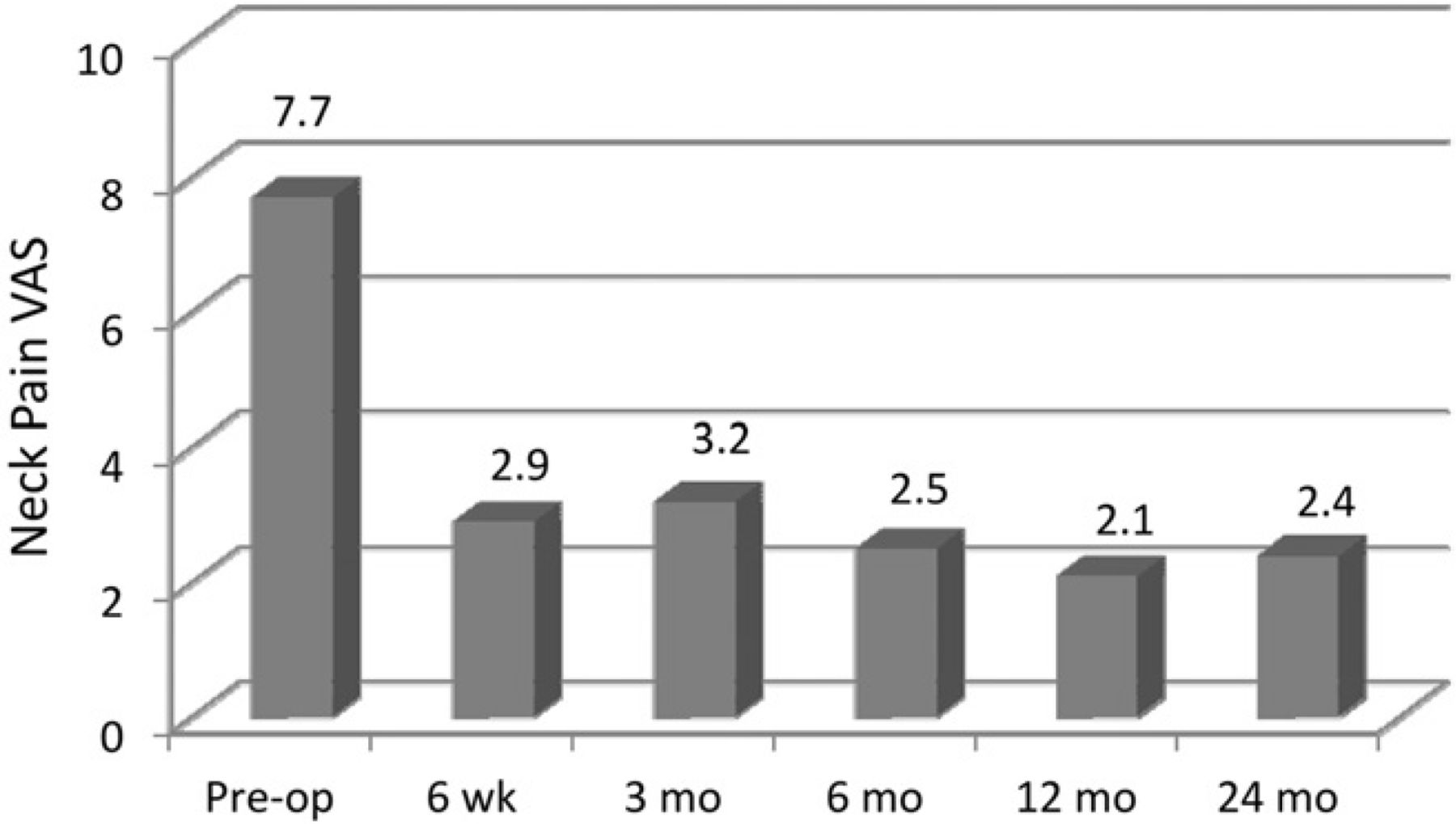
Mean VAS scores for neck pain through follow-up. (Pre-op, preoperatively.)

Mean VAS scores for right and left arm pain. (Pre-op, preoperatively.)
SF-36 Health Survey scores with preoperative versus postoperative comparisons
Though not a primary part of the study, results for 1- versus 2-level TDRs were compared (Table 3). At the 24- month follow-up visit, there were no significant differences between 1- and 2-level patients for change from baseline for NDI (P = .26), right arm pain (P = .97), left arm pain (P = .81), SF-36 PCS score (P = .41), and SF-36 MCS score (P = .091). However, 2-level patients had a significantly greater improvement in reduction in the neck pain VAS score than 1-level patients: 6.3 versus 3.9 (P = .033).
One- and two-level clinical outcomes
Radiographic outcomes
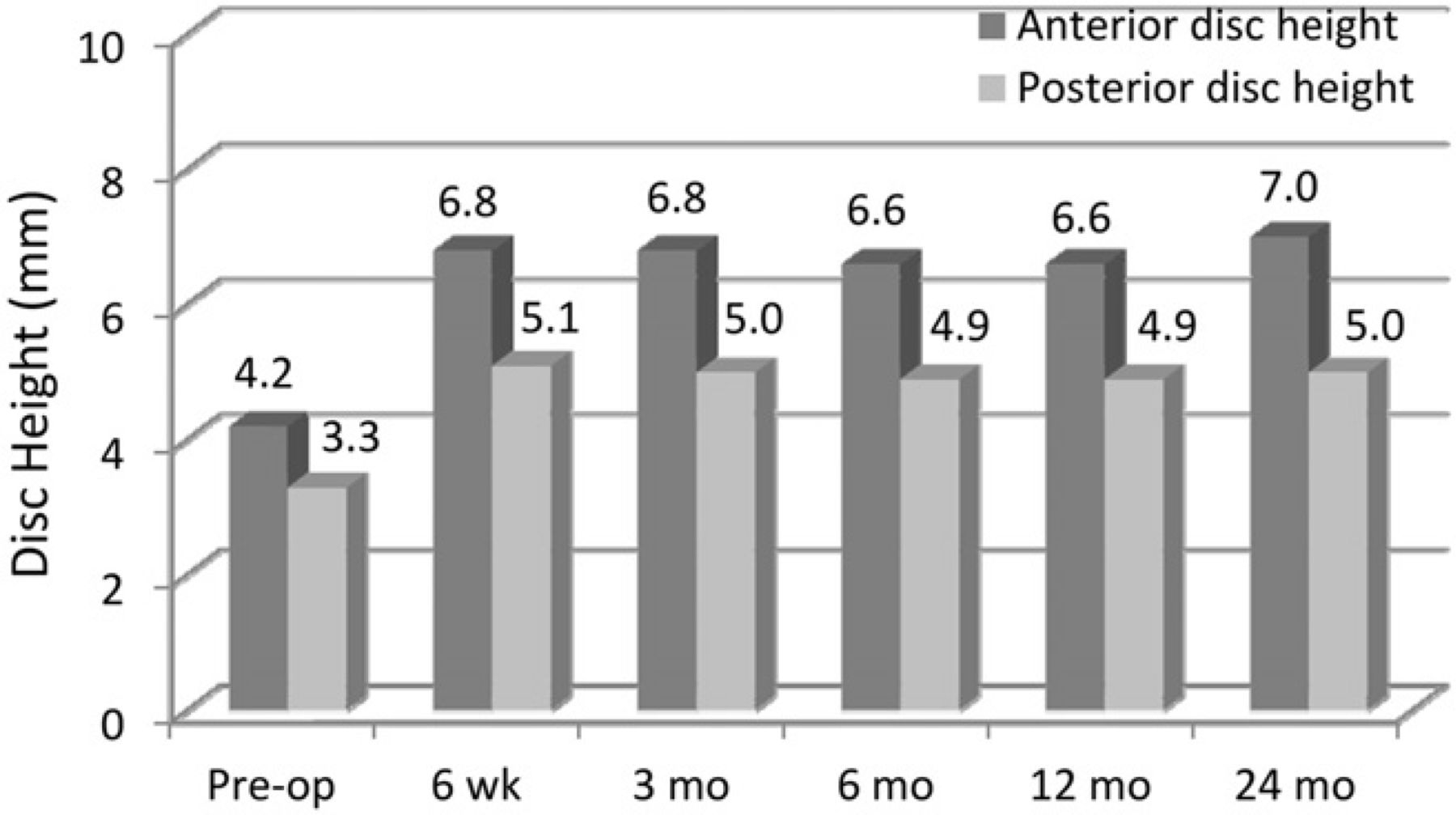
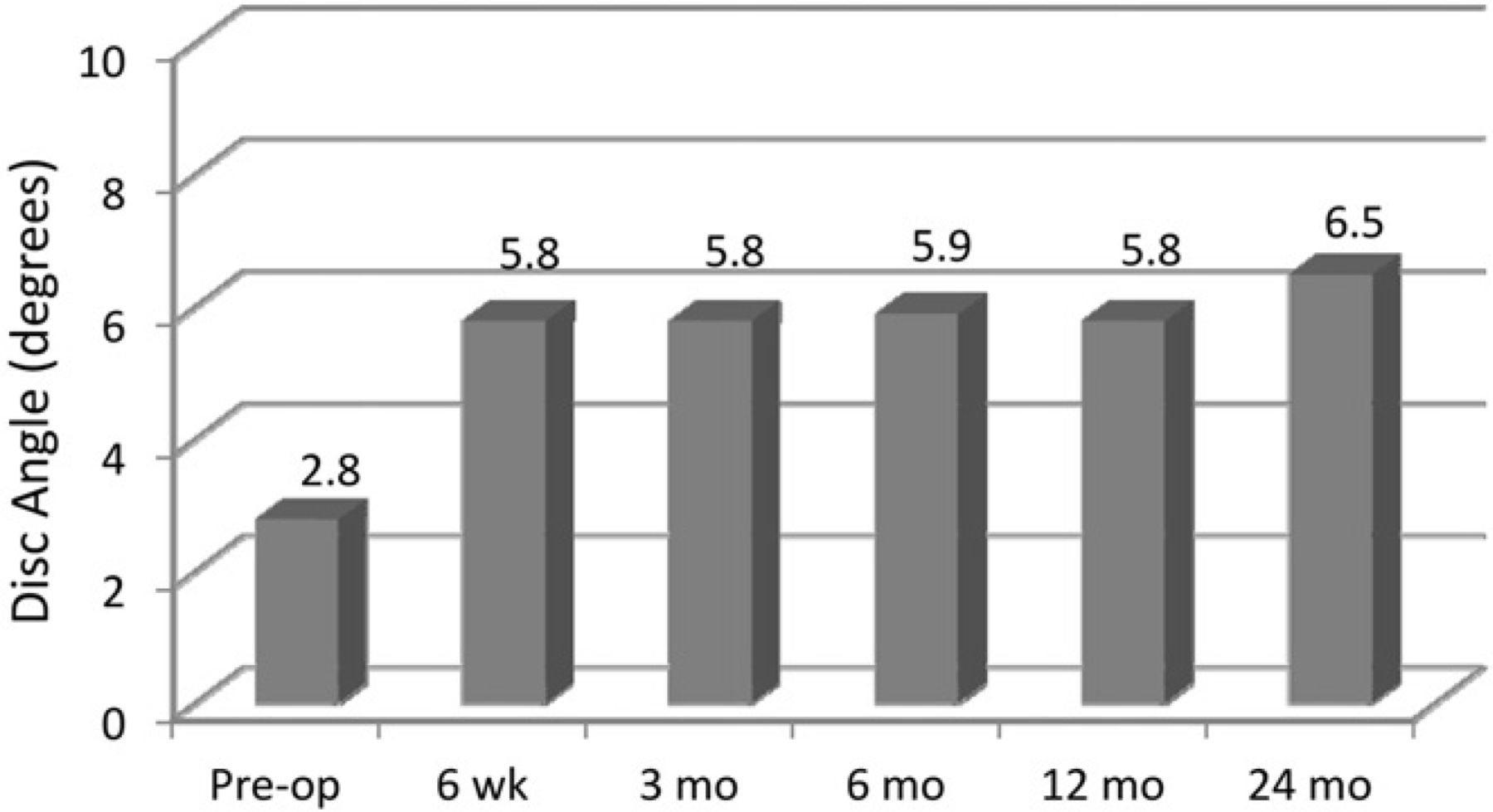
Anterior and posterior disc height increased postoperatively (Fig. 5) and was maintained throughout the 24-month follow-up. The larger increase in anterior disc height contributed to the significant increase in postoperative disc angle (P < .0001) (Fig. 6).
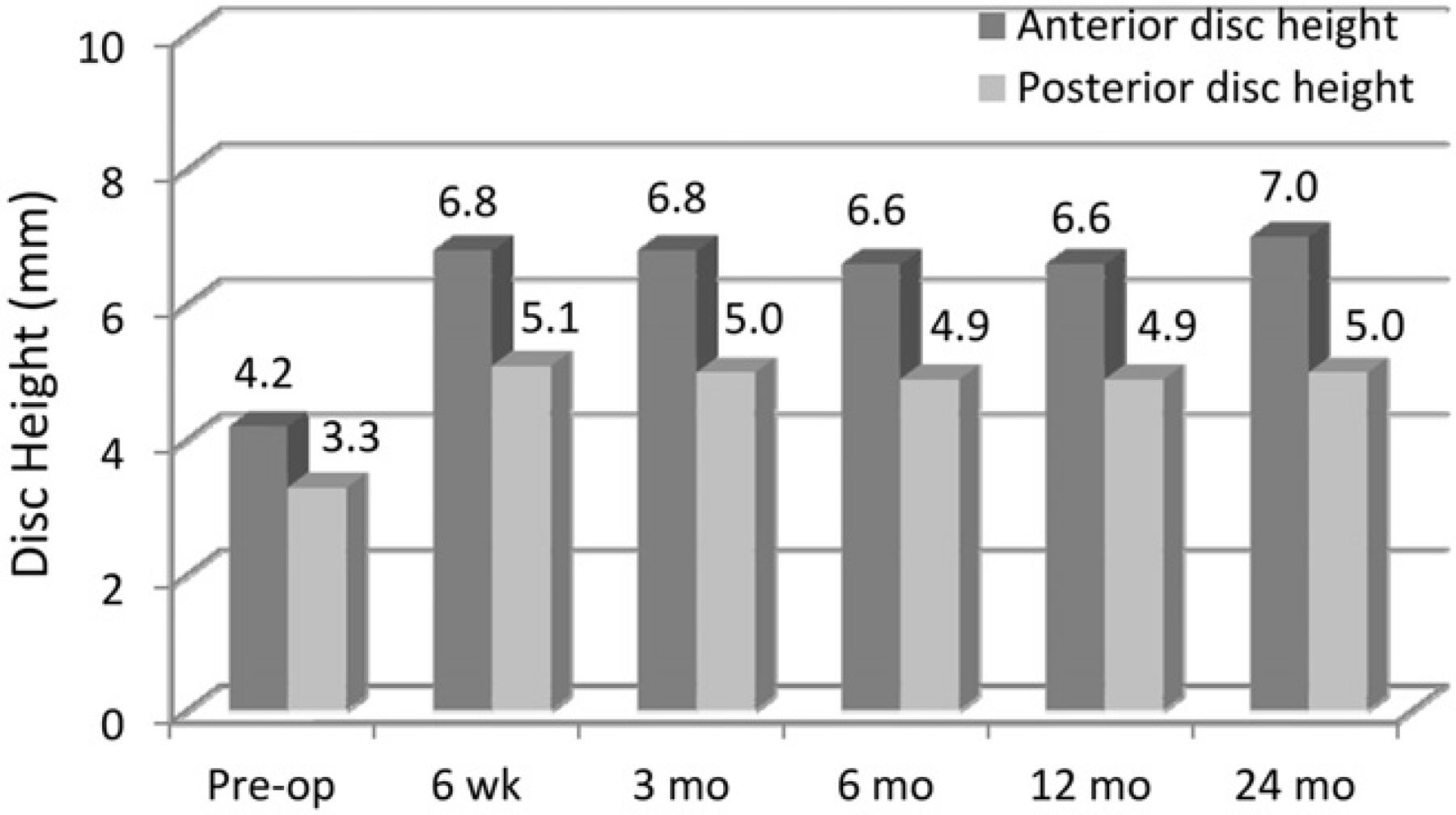
Mean anterior and posterior disc space heights. (Pre-op, preoperatively.)

Mean disc angle preoperatively through follow-up.
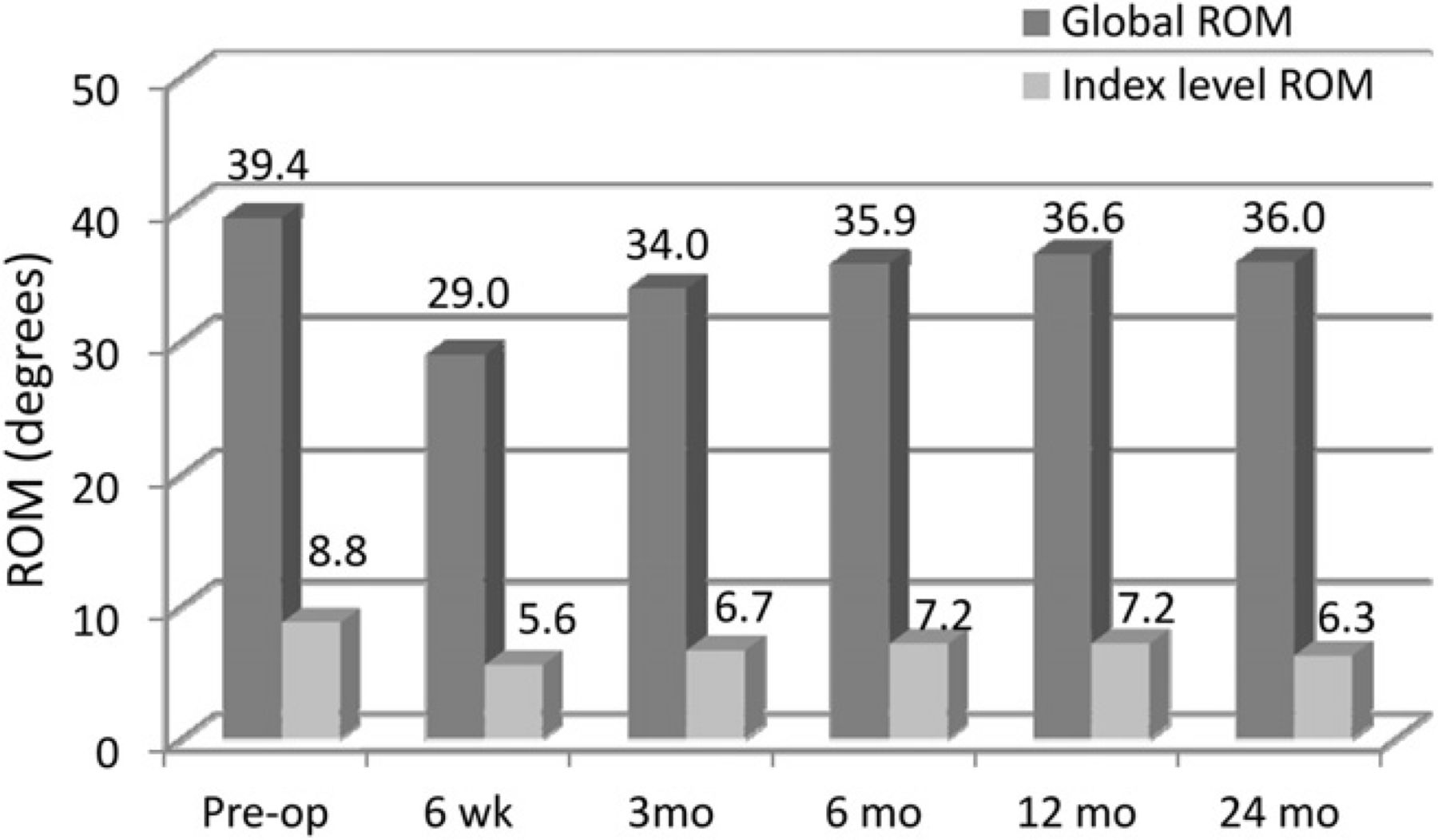
Global and index-level ROM decreased slightly from baseline, but ROM was maintained throughout follow-up (Fig. 7). Lateral bending was also maintained throughout follow-up and was near baseline at 24 months (Fig. 8). Flexion and extension radiographs showing motion of the device are shown in Fig. 9.
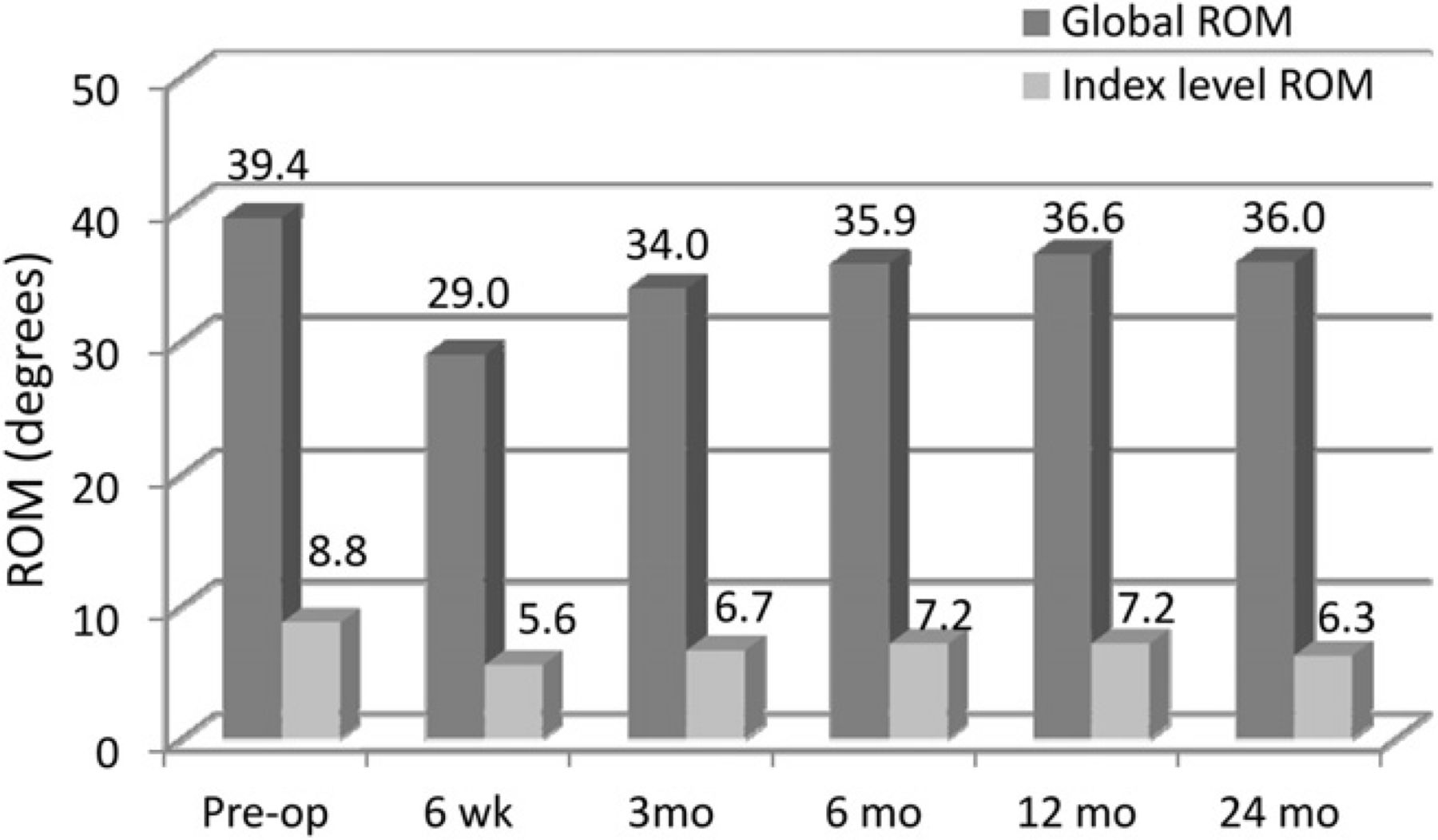
Mean global and index-level ROM. (Pre-op, preoperatively.)

Mean left/right lateral bending of implanted level. (Pre-op, preoperatively.)
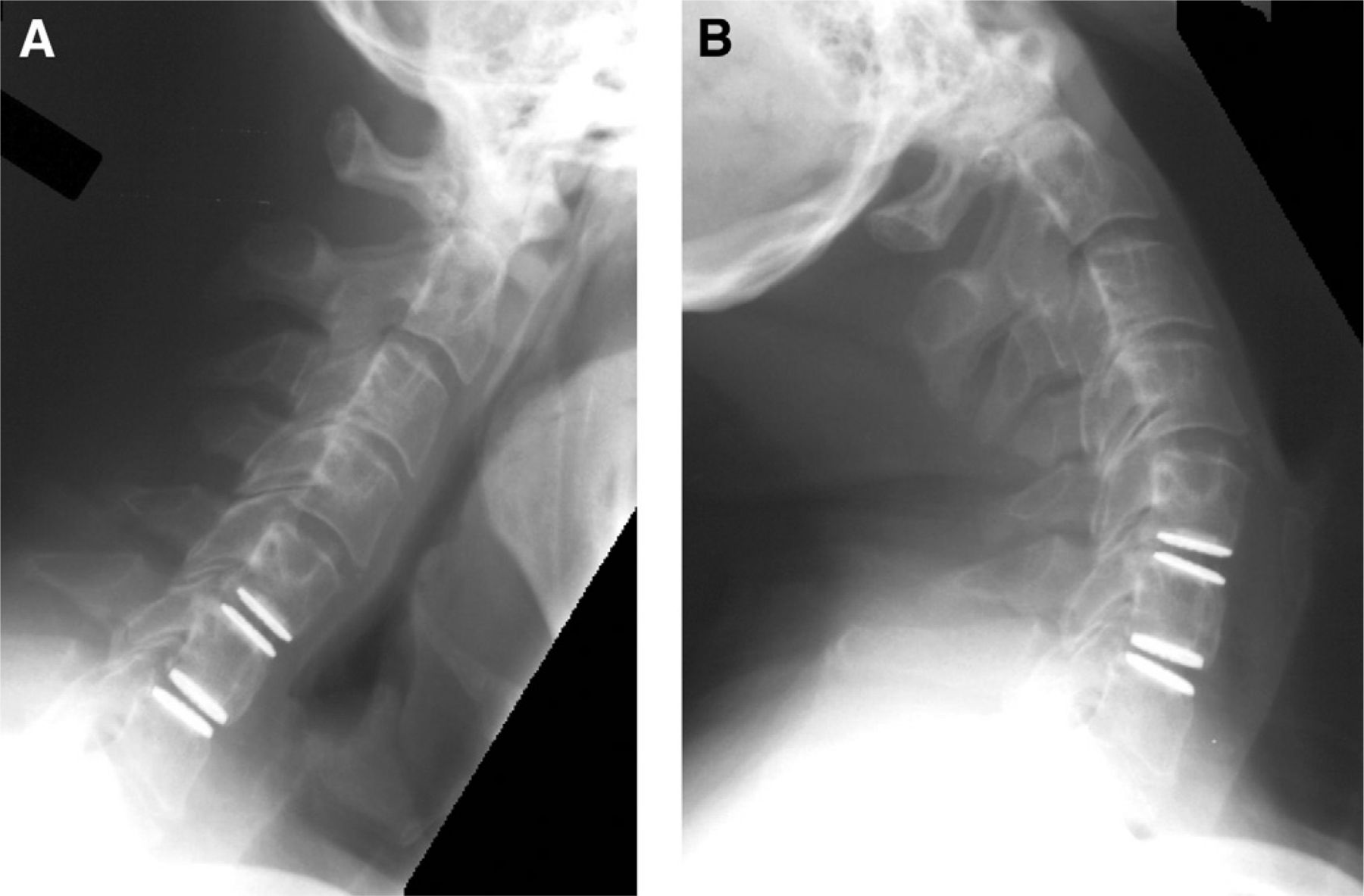
Flexion (A) and extension (B) radiographs of TDRs at C5-6 level with 8.4° of motion and at C6-7 level with 12.4°. The global motion was 39.6°.
Safety outcomes
There were no SAEs related to the device or procedure as adjudicated by the independent clinical events committee. However, 1 subject underwent reoperation for removal of retained wound drain.
Subject overall success
At 12- and 24-month follow-up, 23 subjects (82%) and 24 subjects (89%), respectively, had overall success, with no patients having device removals or revisions, supplemental fixation, or device- or procedure-related SAEs. At 24 months of follow-up, patients with 1-level disc replacement achieved 83.3% success and patients with 2-level replacement achieved 93.3% overall success.
Discussion
The M6-C disc represents a new generation of cTDR design. The compressible core was designed to mimic the compressibility of the natural disc and allows motion with 6 df. Biomechanical testing of this implant found that the kinematic response of the device approximated the response of the intact specimen in flexion-extension.18 It was noted that positioning the implant in the middle versus posteriorly in the sagittal plane did not affect the ROM; however, when the prosthesis was aligned within 1 mm of the disc midline, the center of rotation was most similar to that of the intact spine. The clinical implications of these findings are yet to be defined.
It has been documented through biomechanical testing that maintenance of motion at an implanted segment reduces stresses at adjacent levels.19, 20 Though intuitive, it remains theoretical that this motion results in decreased clinical adjacent-level degeneration. A recent nonrandomized study found no difference in the adjacent segment when comparing cTDR with ACF.21 Other recent prospective randomized studies reported through radiographic evaluation that cTDR reduces the rate of adjacent-segment degeneration compared with ACF.10, 22, 23 These studies investigated artificial discs with a sliding mobile core and domed superior surface. Although such devices should allow more natural motion than a ball-and-socket design, they do not allow motion with 6 df, in contrast to the compressible device in this study.18, 24 Determining whether qualitative motion to more closely mimic natural motion would meaningfully reduce the rate of adjacent-segment degeneration will require a much larger sample size than used in this pilot study.
The results of this prospective FDA-regulated pilot study found that the compressible TDR was associated with significantly reduced neck and arm pain, improved self-reported disability as assessed by the NDI, and improved quality of life as shown by SF-36 Health Survey scores. These results were similar to those reported in a previous study performed outside of the United States using the same device, in which 25 patients with 24-month follow-up showed significant improvement in the NDI, VAS neck and arm pain scores, and the Physical Component Score of the SF-36 Health Survey.25 The NDI scores were also similar to results reported in multiple FDA Investigational Device Exemption trials for other cTDR devices using this same outcome assessment.15 Overall success through 2-year follow-up was achieved in 89% of the patients with no incidences of device- or procedure-related adverse events and no device removal, revision, or supplemental fixation.
With respect to radiographic results, although there was variation in the preoperative and postoperative values in the various studies, the decrease in ROM in early follow-up with increasing values later seen in our study has been noted in another TDR trial.4
In this study 18 patients underwent TDR at 2 levels. The outcomes were not significantly different from the singlelevel cases with the exception of greater improvement in neck pain noted in the 2-level group. There are few prospective data published on 2-level cervical disc replacement. Interestingly, in a prospective, randomized study comparing TDR with ACF in patients with 2-level cervical pathology, both groups improved significantly, with the TDR group having significantly greater improvement in NDI scores.26 To date, only 2 cTDR devices (Prestige LP [Medtronic Sofamor Danek Inc., Minneapolis, MN] and Mobi-C [LDR Spine, Troyes, France]) evaluating 2-level symptomatic disc disease have completed US FDA prospective, randomized studies.13, 27
Limitations of this early-phase feasibility study include a relatively low number of patients without a control group. However, patient selection criteria for this study were very similar to other larger TDR studies, as were the outcomes.
The results of this US FDA–regulated feasibility study suggest that the device studied produced results comparable with other cTDRs. Additional prospective investigation with a larger number of patients and longer follow-up is warranted to establish whether quality of motion with a novel compressible core improves clinical outcome and reduces adjacent-level disease.
- © 2012 Published by Elsevier Inc. on behalf of ISASS - International Society for the Advancement of Spine Surgery.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Related Articles
Cited By...
- Innovation and Adversity in Spine Surgery: A Retrospective
- Current Concepts of Cervical Disc Arthroplasty
- 2021 Position Statement From the International Society for the Advancement of Spine Surgery on Cervical and Lumbar Disc Replacement
- Cervical Disc Arthroplasty: Rationale and History
- Does Resection of the Posterior Longitudinal Ligament Affect the Stability of Cervical Disc Arthroplasty?