


Abstract
Background A feasibility trial was conducted to evaluate the initial safety and clinical use of a next-generation artificial cervical disc (M6-C artificial cervical disc; Spinal Kinetics, Sunnyvale, CA) for the treatment of patients with symptomatic degenerative cervical radiculopathy. A standardized battery of validated outcome measures was utilized to assess condition-specific functional impairment, pain severity, and quality of life.
Methods Thirty-six consecutive patients were implanted with the M6-C disc and complete clinical and radiographic outcomes for 25 patients (mean age, 44.5 ± 10.1 years) with radiographically-confirmed cervical disc disease and symptomatic radiculopathy unresponsive to conservative medical management are included in this report. All patients had disc-osteophyte complex causing neural compression and were treated with discectomy and artificial cervical disc replacement at either single level (n = 12) or 2-levels (n = 13). Functional impairment was evaluated using the Neck Disability Index (NDI). Evaluation of arm and neck pain severity utilized a standard 11-point numeric scale, and health-related quality of life was evaluated with the SF-36 Health Survey. Quantitative radiographic assessments of intervertebral motion were performed using specialized motion analysis software, QMA (Quantitative Motion Analysis; Medical Metrics, Houston, TX). All outcome measures were evaluated pre-treatment and at 6 weeks, 3, 6, 12, and 24 months.
Results The mean NDI score improved from 51.6 ± 11.3% pre-treatment to 27.9 ± 16.9% at 24 months, representing an approximate 46% improvement (P <.0001). The mean arm pain score improved from 6.9 ± 2.5 pre-treatment to 3.9 ± 3.1 at 24 months (43%, P =.0006). The mean neck pain score improved from 7.8 ± 2.0 pre-treatment to 3.8 ± 3.0 at 24 months (51%, P <.0001). The mean PCS score of the SF-36 improved from 34.8 ± 7.8 pre-treatment to 43.8 ± 9.3 by 24 months (26%, P =.0006). Subgroup analyses found that patients treated at single level and those with a shorter duration of symptoms showed better functional results. By 24 months, the mean range of motion (ROM) value at the treated level had returned to approximately pretreatment levels (12.2° vs 11.1°). There were no serious device-related adverse events, surgical re-interventions or radiographic evidence of heterotopic ossification, device migration, or expulsion in this study group.
Conclusions These findings indicate substantial clinical improvement for all function, pain, and quality of life outcomes in addition to maintenance of ROM and increase in disc height at the treated level(s). The findings also exhibit an acceptable safety profile, as indicated by the absence of serious adverse events and reoperations following arthroplasty with a next-generation artificial cervical disc replacement device.
Most patients with symptomatic degenerative cervical radiculopathy realize immediate and sustained clinical benefit from surgical treatment that includes discectomy coupled with osteophyte removal to decompress the nerve roots at the affected level1–3; however, this procedure results in structural alterations that are less than optimal from an anatomic and biomechanical standpoint.3 In fact, despite satisfactory clinical outcomes, cervical discectomy, and neural decompression alone almost always results in disc space collapse.1
To maintain disc height and stability after discectomy, an interbody instrumented fusion procedure is commonly performed. Unfortunately, fusing the affected segment not only diminishes motion at the fused level4 but has an untoward biomechanical effect on adjacent discs that, in essence, must overcompensate for the loss of natural motion in the fused segment.5, 6 The biomechanical modifications that occur after fusion have been shown radiographically to increase the risk of further disc degeneration and osteophyte formation at adjacent levels.7, 8 While debate remains as to the shortterm clinical importance of these radiographic changes,9 it appears that there is a substantial possibility that new disease will develop at an adjacent level over the long term.10, 11
Artificial cervical disc replacement has emerged as a viable treatment alternative to fusion for the management of symptomatic compressive radiculopathies.12, 13 The designs of the first generation artificial cervical discs evolved logically from large joint arthroplasty devices (eg, ball-and-socket),13–15 providing a certain degree of normal motion.16 However, unlike large joints, the movement of the intervertebral disc joint involves complex coupled motions requiring 6 degrees of freedom.17, 18 To date, these complex kinematic properties have been difficult to reproduce.
This single-arm, prospective feasibility study evaluated the preliminary safety and effectiveness of a next-generation artificial cervical disc (M6-C artificial cervical disc; Spinal Kinetics, Sunnyvale, CA) in the treatment of patients with symptomatic cervical radiculopathy. This novel disc system is designed to replicate the anatomic, physiologic, and biomechanical characteristics of the native disc by incorporating a compressible nucleus within a woven fiber annulus. These unique properties allow for natural kinematics including axial compression, translation independent of rotation, and progressive resistance to motion resulting from a physiologically restrained construct. Thus the quality of motion closely mimics that of the native intervertebral cervical disc.
Materials and methods
Patients
The patients enrolled in this study were recruited from the standard cervical spine patient population at the National Institute of Rehabilitation in Mexico City, Mexico. Patients treated presented with clinically and radiographically confirmed symptomatic degenerative cervical radiculopathy, unresponsive to conservative medical management lasting at least 6 weeks. This patient population usually does not present to the Institute of Rehabilitation early in the disease process and is composed primarily of manual laborers who typically do not have insurance or disability benefits. They must work until symptoms reach a level that is intolerable and finally require surgical treatment. In a significant portion of the patient population there is radiographic evidence of disc degeneration at levels adjacent to the proposed surgery level(s). Historically, cervical fusion has been the only surgical option for these patients.
The study was approved by the local Ethics Committee and all patients signed a study specific informed consent. Thirty-six consecutive patients who met specific eligibility criteria, including persistent neurological symptoms associated with cervical radiculopathy by exhibiting at least 1 clinical sign associated with the vertebral level to be treated, moderate functional deficits as indicated by a minimum score of 30% on the Neck Disability Index (NDI), and definitive clinical and radiographic evidence of cervical radiculopathy at C3-7 with or without spinal cord compression as documented on magnetic resonance imaging (MRI), were enrolled in the study. Radiographic diagnosis of neural compression included evidence of disc-osteophyte complex. Patients were excluded if they exhibited advanced degenerative changes (eg, spondylosis) at the index vertebral level as evidenced by bridging osteophytes, average range of motion (ROM) <4°, disc height <20% of the anteroposterior (AP) width of the inferior vertebral body (as measured on the lateral view), subluxation >3 mm, or kyphotic deformity >20° on neutral radiographs. The study was initiated with a final follow-up at 12 months post-surgery; however, the study protocol was amended during the follow-up time period to include an additional 24-month follow-up. Twenty-five patients were able to return to the clinic for complete clinical and radiographic evaluations at the 24-month follow-up time point. Although the remaining 11 patients do not have complete 24-month clinical and radiographic evaluations, they are being followed by the clinic and are included in the evaluation of device safety.
Clinical outcomes
Patient outcomes were measured prior to surgery and at 6 weeks and 3, 6, 12, and 24 months post-procedure. Functional impairment was evaluated with the Neck Disability Index (NDI),19 arm and neck pain severity was evaluated with a standard 11-point numeric scale,20 and health-related quality of life was evaluated using the SF-36 Health Survey.21 To evaluate neurological function, a standard cervical neurological examination including sensation, motor function, and reflexes was performed preoperatively and at all postoperative time points.
Furthermore, all patients underwent a series of plain film x-rays including AP, lateral, flexion-extension, and right and left lateral bending preoperatively and at the postoperative evaluations. Intervertebral ROM at the treated level, global ROM for the entire cervical spine, and disc height were determined independently using proprietary quantitative imaging software (QMA; Medical Metrics, Houston, TX).22, 23 Additionally, postoperative x-rays were evaluated to assure proper placement of the device and to identify evidence of heterotopic ossification (HO).
Surgery
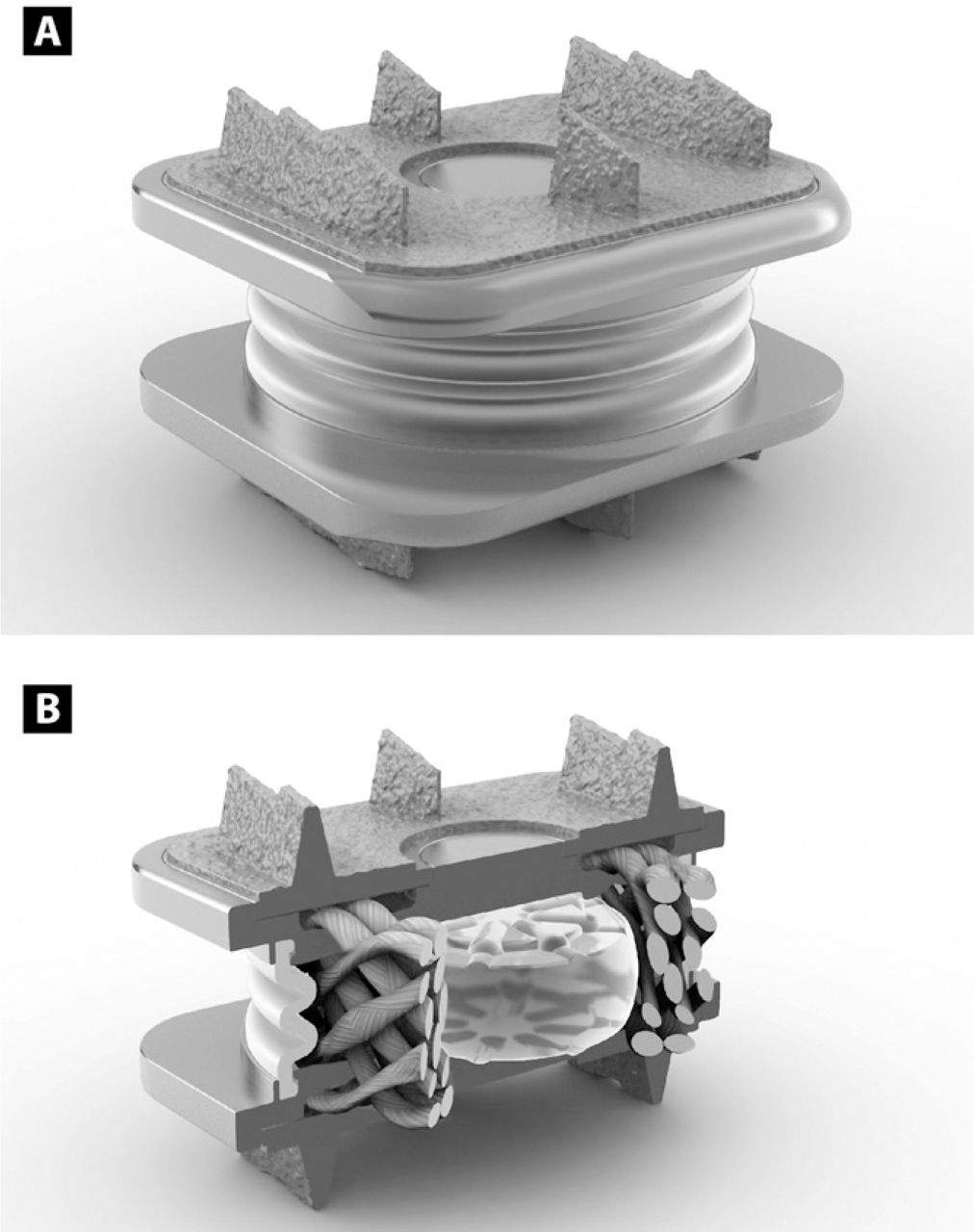
All patients were implanted with the M6-C artificial cervical disc (Spinal Kinetics, Sunnyvale, CA) (Fig. 1). It is designed to replicate the anatomic structure of a natural disc by incorporating an artificial nucleus and annulus. The compressible polymer nucleus of the artificial disc is designed to simulate the function of the native nucleus, while the surrounding multi-layer high tensile strength fiber annulus is intended to provide progressive resistance to motion and a controlled range of motion. The fibers of the annulus are attached to titanium endplates in a unique manner that allows the physiologic angles of motion in flexion, extension, lateral bending, and axial rotation as well as compression. The device also has a polymer sheath encasing the core and fiber construct to inhibit tissue in-growth as well as capture potential wear debris. The endplates are attached to the vertebral body via 3 keels on the superior and inferior surfaces and are coated with porous titanium to increase bone-contact surface area and provide acute fixation to the superior and inferior vertebral bodies within the intervertebral space.

(A) Graphical illustration of the artificial cervical disc (M6-C artificial cervical disc, Spinal Kinetics, Sunnyvale, CA). (B) Cut-away image of the disc illustrating a compressible polymer core to simulate the nucleus surrounded by woven fibers arranged circumferentially to simulate the annulus.
The unique design of the current device allows all 6 degrees of freedom, including angular motion in flexion-extension, lateral bending, and axial rotation as well as allowing independent translations along the 3 anatomic planes (anterior-posterior, side to side, and axial compression).
The device is implanted using a standard anterior approach after the target disc space is identified and confirmed via fluoroscopy. Distractor pins, intervertebral distracters, and spreaders are used and patients undergo a complete discectomy to the posterior longitudinal ligament and uncovertebral junction. Osteophytes are removed as needed to assure parallel endplates and adequate surface to accept the device, but excessive removal of bone is not suggested. Device specific trial implants are provided to determine the appropriate size and position of the device. After determining proper device size and position, a procedure-specific chisel is used to create keel tracks in the superior and inferior vertebral bodies. The device is then loaded on the implant inserter and tapped into the desired position in the disc space.
All patients were advised in standard postoperative precautions and prescribed a nonsteroidal anti-inflammatory agent (ketorolac) for 10 –14 days post-surgery, in an effort to minimize the occurrence of heterotopic ossification.
Statistical methods
Background characteristics and clinical results are presented as descriptive statistics or frequency and percentage distributions, as appropriate. Baseline values for all outcomes were compared only with 24-month follow-up values using the paired t test; 2-tailed and individual significance values are presented for each outcome in lieu of adjusting the individual significance levels for multiple comparisons.
Results
Patient characteristics
Thirty-six patients were implanted with the device in the initial M6-C artificial cervical disc feasibility study. This report describes the clinical findings for 25 patients who have complete 24-month clinical and radiographic follow-up with all 36 patients included in the evaluation of safety by the monitoring of adverse events. Eighteen patients (72%) were actively employed as manual laborers at the time of surgery and the mean duration of symptoms was 31.8 months, which is uncharacteristically long for this patient population. There were 12 single level cases and 13 2-level cases, representing 38 treated levels. A wide range of conservative interventions were attempted unsuccessfully prior to enrollment. Statistical summary results for background characteristics of this study group are provided in the Table.
Patient baseline characteristics
Clinical and radiographic outcomes
Significant improvement in functional impairment measured by the NDI was present within 6 weeks of surgery and was sustained through 24 months of postoperative follow-up (Fig. 2). Overall, the mean NDI score improved from 51.6 ± 11.3% pre-treatment to 27.9 ± 16.9% at 24 months. The average decrease was 23.2 ± 17.6% and the corresponding mean percentage improvement in NDI was approximately 46% (P <.0001) at 24 months.
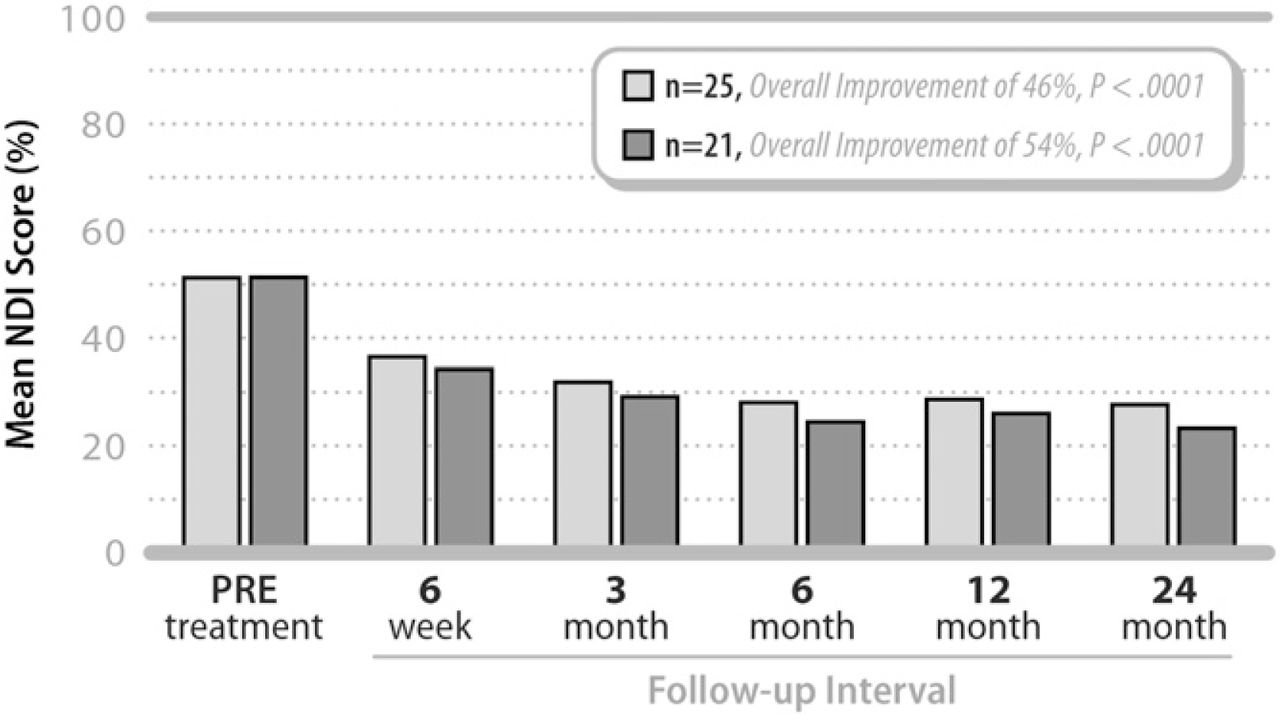
Mean Neck Disability Index (NDI) values pretreatment and at each follow-up interval for all patients (N = 25) as well as for patients with ≤48 months of symptoms (n = 21).
A scatterplot comparing change in NDI with duration of preoperative symptoms suggested a potential relationship, with notably worse clinical results, among patients with symptoms greater than 48 months prior to surgery. Subsequently, in a subgroup analysis of the 21 patients with symptoms of less than 48 months, the mean NDI value improved from 51.8 ± 11.9% pre-treatment to 23.7 ± 14.7% at 24 months, representing an average change of 27.6 ± 15.7 percentage points (54%, P <.0001) (Fig. 2).
NDI results were also better for single level patients than for 2-level patients. The average decrease at 24 months for single level patients was 25.5 ± 18.3 percentage points or approximately 52% (P =.001), whereas the average decrease for 2-level patients was 21.3 ± 17.5 percentage points or approximately 38% (P =.0009).
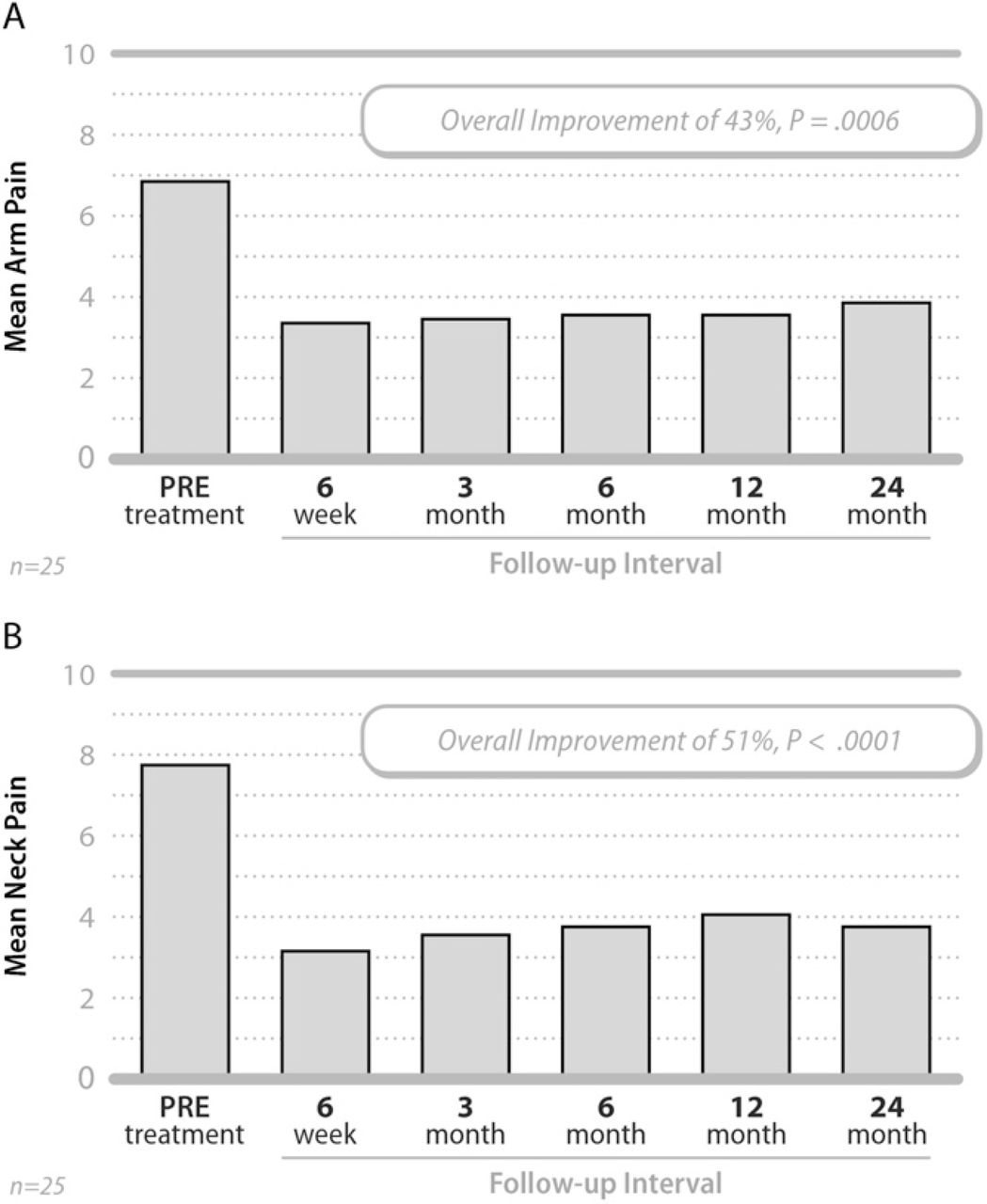
Patients also experienced improvements in arm and neck pain severity, beginning at 6 weeks and continuing through 24 months post-surgery (Fig. 3). The mean arm pain score improved from 6.9 ± 2.5 pre-treatment to 3.9 ± 3.1 at 24 months, an average change of 3.0 ± 3.8 points or approximately 43% (P =.0006). The mean neck pain score improved from 7.8 ± 2.0 pre-treatment to 3.8 ± 3.0 at 24 months, representing an average change of 4.0 ± 3.7 points or approximately 51% (P <.0001).
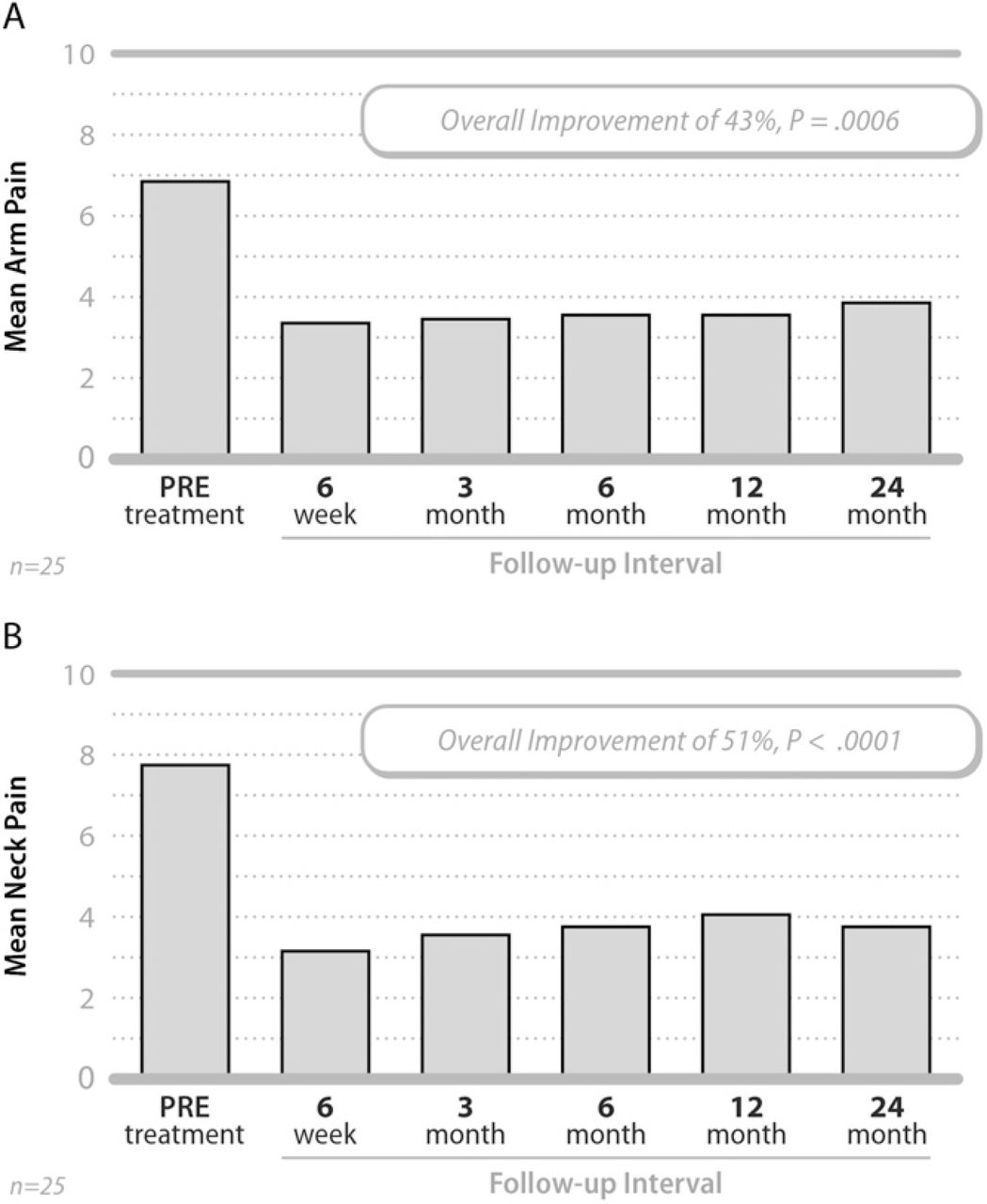
Mean arm (A) and neck (B) pain severity values pretreatment and at each follow-up interval.
As with the NDI, patients treated at a single level had somewhat better improvement in pain severity than those treated at 2 levels. For example, the average decrease at 24 months for single level patients was 3.7 ± 3.6 points (50%, P =.004) and 4.0 ± 3.7 points (51%, P =.003) for arm and neck pain, respectively; whereas the average decrease for 2-level patients was 2.4 ± 4.2 points (38%, P =.06) and 4.0 ± 3.9 points (43%, P =.003) for arm and neck pain, respectively.
The mean physical component summary (PCS) score of the SF-36 improved from 34.8 ± 7.8 pre-treatment to 43.8 ± 9.3 by 24 months, reflecting an average change of 8.9 ± 11.0 points (26%, P =.0006). The mean mental component summary (MCS) score improved from 42.6 ± 11.1 pre-treatment to 48.6 ± 9.6 by 24 months, reflecting an average change of 5.5 ± 10.7 points (14%, P =.02).
By 24 months, the mean ROM value at the treated level had returned to approximately pretreatment levels (ie, 12.2° vs 11.1°) and global ROM was essentially unchanged over the same period (ie, 45.3° vs 45.6°) (Fig. 4). Mean disc height improved from 3.1-mm pre-treatment to 5.4 mm at 24 months (Fig. 5).
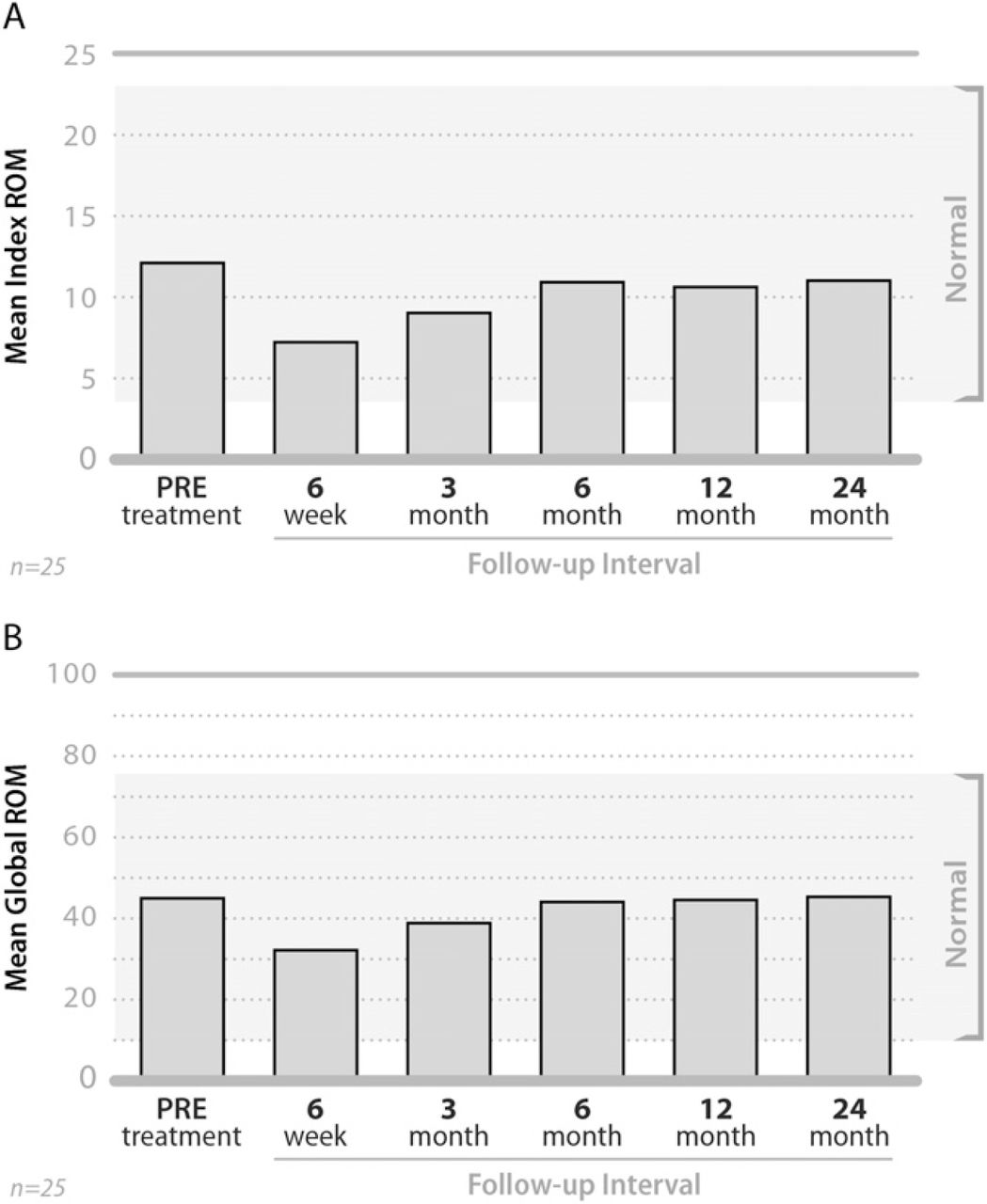
Mean ROM values at the treated level (A) and globally for the entire neck (B) pretreatment and at each follow-up interval.

Mean disc height values pretreatment and at each follow-up interval.
Adverse events
If there was an untoward event that was considered serious in nature and related to the device or device surgery, resulted in a secondary surgical intervention to remove, modify or replace the original device at the treated level, or was the result of device migration or expulsion, the patient was to be considered an adverse event failure. There have been no serious device-related adverse events, surgical re-interventions, or radiographic evidence of heterotopic ossification, device migration, or expulsion resulting in patient failure (Fig. 6).

Representative lateral radiographs illustrating a single level (A) and a 2-level (B) cervical disc replacement.
Discussion
Restoring normal biomechanical function to a diseased cervical motion segment requires a total disc replacement device that mimics inherent disc kinematics. Most of the first-generation artificial cervical discs fail to fully replicate the normal visco-elastic disc structure, lacking internal elastic stiffness or restraint and axial compressibility.18 First-generation artificial cervical discs deprived of the full 6 degrees of freedom intrinsic to the native disc may not provide maximum clinical benefit to the patient, whereas the current device does have that capability.
Although this trial did not have an active control group, the study utilized a standardized battery of validated outcome measures to assess functional impairment, pain severity, quality of life, and radiographic assessment similar to the outcomes measures of larger previously published cervical artificial disc studies. Due to the relatively small sample size of the current feasibility study, caution should be used in comparing these results with those of the larger studies. The average improvement in clinical outcomes in this study are somewhat less than those reported in larger, randomized comparative studies for other cervical discs. For example, the mean NDI improvement in this trial is 23.2% over 24 months compared to the results from 3 randomized controlled IDE trials in the US: 36.0% for the Prestige,24 28.9% for the Bryan,25, 26 and 33.0% for the ProDisc-C.27, 28 However, the mean improvement in NDI among the subgroup of patients in the current study with the duration of symptoms of less than 48 months (27.6 percentage points) compares more favorably with the results from those 3 studies.
Also to be taken into consideration is the fact that in this study patients were treated at both single level and 2-level, with clinical results somewhat less for the 2-level cases. This served to dampen the overall magnitude of treatment effect. Nonetheless, it is not clear whether the number of levels treated is an independent predictor of treatment success after total artificial disc arthroplasty. Contradictory to our results, Pimenta et al29 reported the opposite trend with patients treated at 2-level having better clinical outcomes than single level cases. On the other hand, Goffin et al30 failed to note any clinical differences between single level and 2-level artificial disc replacement cases.
The socioeconomic and sociocultural makeup of our patient population may also have had an impact on results and deserves consideration in interpreting the findings. This patient population included mostly females (92%) with an uncharacteristically long mean duration of symptoms of 31.8 months. Eighteen patients (72%) were actively employed as “manual laborers” at the time of surgery with the need to return to work within the shortest possible/tolerable time. Given that fact, it is likely that this patient population is at risk for work-related re-injury and/or progressive spinal degeneration at adjacent levels, causing recurrent or new onset symptoms. Despite this difficult-to-treat patient population, we are very encouraged with the results.
All patients treated in this study were prescribed nonsteroidal anti-inflammatory drugs for 10 –14 days post-procedure, in an effort to minimize the occurrence of heterotopic ossification. Independent radiographic evaluation of postoperative x-rays showed no evidence of HO formation at 24 months. In contrast, 2 European studies reported heterotopic ossification rates of 17.8% and 66% associated with implantation of the Bryan31 and ProDisc-C32 artificial discs, respectively. Unintended fusion (ie, heterotopic ossification resulting in bridging trabecular bone) also occurred in 3 ProDisc-C patients (2.9%) in the US investigational device exemption ProDisc-C randomized trial.27
Conclusions
The 24 month results of the initial M6-C artificial cervical disc clinical trial indicate progressive, substantial clinical improvement over time in function, pain, and quality of life, in addition to maintenance of ROM and improvement in disc height at the treated level(s). This device also exhibits an excellent safety profile as evidenced by the absence of serious device-related adverse events, surgical reinterventions, or radiographic evidence of device migration or expulsion. In addition, no patients in this study exhibited HO at 24 months post-surgery. This finding seems to indicate that the use of post-surgery NSAIDS minimizes the occurrence of heterotopic ossification.
This artificial cervical disc represents an evolution of previous disc systems, which results in an advancement of cervical disc technology. The current device is intended to replicate the anatomical structure of the native disc, incorporating all 6 degrees of freedom in its kinematics profile. The novel design results in a physiologically restrained construct with progressive resistance to motion.
Acknowledgments
This study was supported by Spinal Kinetics, Inc. (Sunnyvale, CA). The authors appreciate the manuscript assistance, statistical guidance and data analysis provided by Jon E. Block, Ph.D., Harrison A. Stubbs, Ph.D. and Phillip Colla.
- © 2010 SAS - The International Society for the Advancement of Spine Surgery. Published by Elsevier Inc. All rights reserved.
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License, permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}