


Abstract
Background Progenitor cells derived from intervertebral disc tissue demonstrated immunomodulatory and regenerative properties in preclinical studies. We report the safety and efficacy results of a US Food and Drug Administration–approved clinical trial of these cells for the treatment of symptomatic degenerative disc disease.
Methods Sixty patients with symptomatic single-level lumbar degenerative disc disease (mean age 37.9 years, 60% men) were enrolled in a randomized, double-blinded, placebo-controlled Phase I/Phase II study at 13 clinical sites. They were randomized to receive single intradiscal injections of either low-dose cells (N = 20), high-dose cells (N = 20), vehicle alone (N = 10), or placebo (N = 10). The primary endpoint was mean visual analog scale (VAS) pain improvement >30% at 52 weeks. Disc volume was radiologically assessed. Adverse events (AEs), regardless of whether they were related to treatment, were reported. Patients were assessed at baseline and at 4, 12, 26, 52, 78, and 104 weeks posttreatment.
Results At week 52, the high-dose group had a mean VAS percentage decrease from baseline (−62.8%, P = 0.0005), achieving the endpoint of back pain improvement >30%; the mean change was also significantly greater than the minimal clinically important difference of a 20-point decrease (−42.8, P = 0.001). This clinical improvement was maintained at week 104. The vehicle group had a smaller significant decrease in VAS (–52.8%, P = 0.044), while the low-dose and placebo groups showed nonsignificant improvements. Only the high-dose group had a significant change in disc volume, with mean increases of 249.0 mm3 (P = 0.028) at 52 weeks and 402.1 mm3 (P = 0.028) at 104 weeks. A minority of patients (18.3%) reported AEs that were severe. Overall, 6.7% of patients experienced serious AEs, all in the vehicle (n = 1) or placebo (n = 3) groups, none treatment related.
Conclusions High-dose allogeneic disc progenitor cells produced statistically significant, clinically meaningful improvements in back pain and disc volume at 1 year following a single intradiscal injection and were safe and well tolerated. These improvements were maintained at 2 years post-injection.
Level of Evidence 1.
Clinical Trial Registration NCT03347708—Study to Evaluate the Safety and Preliminary Efficacy of Injectable Disc Cell Therapy, a Treatment for Symptomatic Lumbar Intervertebral Disc Degeneration.
Introduction
Chronic low back pain (CLBP) affects 12% to 30% of US adults at a given time.1 It is a leading cause of disability worldwide and the leading cause of years lived with disability in developing countries.2,3 The 2010 Global Burden of Disease Study estimated that lower back pain is among the top 10 diseases and injuries that account for the highest number of disability-adjusted life years worldwide.4
Low back pain often arises from degeneration of the intervertebral disc. As the world population ages, CLBP will increase substantially due to the deterioration of the intervertebral discs in older people.5 There is evidence, however, that if CLBP starts at a younger age, it is likely that the pain is discogenic in nature.6 Discogenic pain is pain arising from a damaged intervertebral disc, particularly associated with degenerative disc disease (DDD), a chronic and progressive condition that is characterized by the breakdown of tissue and inflammation within the intervertebral disc, resulting in pain and disability. The intervertebral disc is relatively avascular, hypoxic, and hypocellular, making it perhaps more prone to degenerative conditions.7
Disc degeneration is associated with structural changes within the disc, including loss of hydration, decrease in cell volume, and diminished extracellular matrix molecules (eg, proteoglycans). These degenerative changes likely contribute to disc tissue becoming less functional and can result in a significant volumetric decrease in the intervertebral disc.8 As a consequence, patients can develop pain and disability.
Degenerated discs are characterized by upregulation of inflammatory cytokines, such as interleukins and tumor necrosis factor alpha,9 which can lead to further tissue breakdown and pain. The breakdown of tissue within the disc is due to the depleted capacity of local cells to produce new extracellular matrix, as well as to an imbalance in anabolic and catabolic activity within the tissue.7 Other aspects of a pathologic symptomatic disc include abnormal innervation, devascularization, and changes to the disc endplates. These changes can result in global tissue structural damage that may be manifested by acute and chronic pain and may eventually result in structural failure.
The pain and disability associated with symptomatic DDD can have significant short- and long-term consequences ranging from broad to highly specific social, physical, and economic impacts. At the macro level, the annual expenditure to treat CLBP in the United States is estimated to be over $100 billion,3 creating a significant burden on the economy and the patients dealing with CLBP. DDD not only can impair an individual’s ability to perform simple self-care activities and preclude them from enjoying leisure activities, but it can also prevent them from working and contributing meaningfully to society.10
Physically, DDD creates structural changes that result in abnormal mechanical and biological-pathological processes. This can lead to facet joint arthritis, osteophyte formation, spinal canal stenosis, and spondylolisthesis. Current treatment options for symptomatic DDD in early stages are limited. Treatment modalities may include various medications, physical therapy, epidural steroid injections, acupuncture, chiropractic care, and other nonsurgical modalities. When the disease progresses to more advanced stages, is unresponsive to nonoperative measures, and remains clinically debilitating, surgical intervention is indicated. Surgical intervention typically consists of obliteration of the pathological disc followed by fusion or total disc replacement. These surgeries are substantial, costly procedures with significant risks and relatively limited success rates, which should be reserved as a last-resort treatment. Fusions may also result in subsequent adjacent-level degeneration, which has reported incidence rates of up to 62.5% following lumbar fusion surgery.11,12
Given the drawbacks to surgical treatment of symptomatic DDD, a less invasive treatment option is desirable. Allogeneic progenitor cells derived from adult human intervertebral disc tissue have demonstrated both immunomodulatory and regenerative properties in preclinical animal studies, including the ability to increase aggrecan and collagen production and restore disc volume and histology.13 Injectable disc cell therapy (IDCT or rebonuputemcel) is an allogeneic injectable discogenic progenitor cell therapy intended for patients experiencing pain caused by early to moderate DDD. The active ingredient of IDCT is a live disc progenitor cell population derived from the intervertebral disc tissue of adult organ donors that is enriched and expanded in the laboratory into discogenic cells. The discogenic cells are mixed with a viscous sodium hyaluronate solution and excipients to generate IDCT, the final drug product. Preclinical in vitro and in vivo evaluations of discogenic cells have been previously reported and have shown that the cells produce an extracellular matrix that may rebuild the depleted tissue within degenerating discs while not posing any significant safety concerns.13
In the present study, we report the findings of a US Food and Drug Administration (FDA)–approved randomized, double-blinded, vehicle- and placebo-controlled, multicenter combined Phase I and Phase II study to evaluate the safety and tolerability of a single injection of IDCT. Clinical and radiographic outcomes were assessed in human subjects with single-level, symptomatic, early-to-moderate lumbar intervertebral disc degeneration. This is an investigational new drug following a Biologics License Application pathway.
Methods
This study was conducted in accordance with the protocol and consensus ethical principles derived from international guidelines. The protocol, protocol amendments, informed consent form, investigator brochure, and other relevant documents (eg, advertisements) were submitted to an Institutional Review Board (IRB)/Independent Ethics Committee by DiscGenics, Inc., the study sponsor, and reviewed and approved by the IRB/Independent Ethics Committee (Advarra IRB, assurance #00023875) before the study was initiated. Each investigator site also received IRB approval before beginning participation, and informed consent was obtained from all study participants. The clinical trial was registered at Clinical Trials.gov, #NCT03347708.
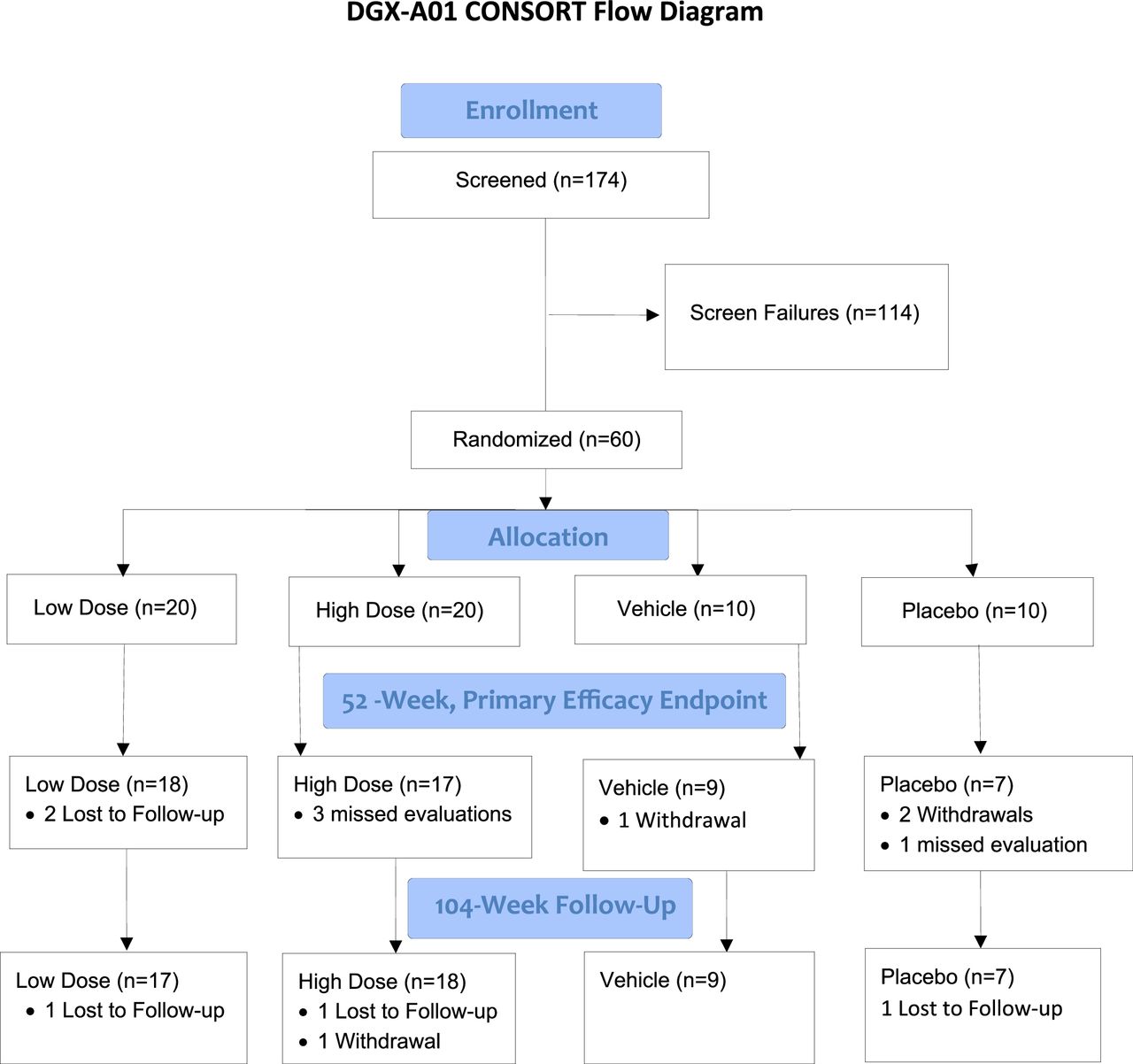
The study was a Phase I/II, first-in-human, randomized, double-blinded, vehicle- and placebo-controlled, multicenter study in patients with single-level, symptomatic lumbar (L3–S1) intervertebral disc degeneration. Although this was first submitted to the FDA as a Phase I study to be followed by a Phase II, at the FDA’s suggestion, the 2 phases were merged so that the evaluation of efficacy endpoints occurred simultaneously with dose escalation and safety outcomes. Patients were followed for a total of 2 years. The CONSORT (Consolidated Standards of Reporting Trials) guidelines were used in the reporting of this study.14
Patients
Thirteen sites in 12 states recruited and treated patients. Sixty patients were enrolled between March 2018 and February 2022. Inclusion and exclusion criteria are listed in Table 1. The sample size was based on visual analog scale (VAS) data (the primary efficacy measure) collected from a pilot study (http://www.spinalrestoration.com/clinical_trials/); within the limitations of a 2-dose and 2 controls early phase trial, size was calculated for a 5% level of significance 1-sided test and 80% power to reject the null hypothesis that mean reduction from baseline VAS score would be equal to 30% when the alternative, mean reduction from baseline greater than 30%, was true. This yielded a sample size of 13 patients for each of the 2 test treatment groups, with the use of a 2:2:1:1 allocation ratio for the 2 cell dose groups, the vehicle group, and the placebo group. Allowing for a 30% dropout rate, a total of 60 patients were randomized: 20 to each cell treatment dose group and 10 each to the vehicle and placebo control groups.
Inclusion and exclusion criteria.
The baseline disease characteristics of the study population were representative of an adult population with treatment-resistant low back pain secondary to lumbar disc degeneration. Median time since symptomatic DDD diagnosis was 5.6 months (range 0.4 [history but no formal diagnosis until the screening visit] to 191.9 months), and there was a prevalence of prior epidural injection (28.3%), physiotherapy (26.7%), insomnia (20.0%), chiropractic procedure (18.3%), and pain in extremity (16.7%). Patients also reported drug hypersensitivity (21.7%), anxiety (16.7%), depression (16.7%), and seasonal allergies (16.7%).
Patients were randomly assigned to 1 of 4 groups: low-dose IDCT (N = 20; 3,000,000 cells/mL), high-dose IDCT (N = 20; 9,000,000 cells/mL), vehicle alone (N = 10; sodium hyaluronate), and placebo (N = 10; normal saline). The dose levels were based on prior rabbit studies, with cell dose increased by the factors resulting from a ratio of average human disc volume to average rabbit disc volume.13 Demographic characteristics were generally balanced between treatment groups, although the median age in the vehicle group was slightly higher than the other groups. Overall patient follow-up was 85.0% at 2 years (see Figure 1, CONSORT flow chart). Demographics and baseline characteristics for all patients and by group are shown in Table 2. Overall, ages ranged from 18 to 61 years. A majority were men (60%) and white (85%). The majority had a modified Pfirrmann score of 4 (65%). Two-thirds of the patients were treated at the L5-S1 level. Although there was some variability within and between groups, differences between groups were not statistically significant.

Study CONSORT (Consolidated Standards of Reporting Trials) flow chart.
Demographics and baseline characteristics for all patients and by treatment group.
Product Description
Discogenic cells are created through a multistep manufacturing process resulting in significant proliferation and phenotypic changes to the cells. This process has been previously described.13 Human disc tissue is obtained from adult organ donors and washed, and unwanted tissue is dissected away. The cells are isolated from the tissue, seeded at 10,000 cells/cm2, expanded using a cocktail of supplements, passaged, and then grown in suspension culture. At the completion of the process, the discogenic cells are subjected to extensive testing prior to use, including identity, purity, potency, and safety evaluations. For this study, treatment was generated from 3 manufactured lots from a single donor.
Discogenic cells possess unique properties that characterize them as progenitor cells. The cells express the surface markers of differentiation to include CD73, CD90, and human leukocyte antigen (HLA)-ABC but do not express CD24, CD34, or HLA-DR/DQ/DP.13,15 This surface marker pattern is tested for and must be present on every batch of discogenic cells. Discogenic cells are multipotent for mesenchymal lineages, including having a strong chondrogenic potential. Endogenously, the cells generate proteoglycan and collagen types 1 and 2, which are the extracellular matrix molecules that make up part of the intervertebral disc.
Materials
Efficacy
For primary efficacy evaluation, patients recorded their perceived low back pain level using a VAS ranging from “no pain” (score of 0) to “pain as bad as it could be” or “worst imaginable pain” (score of 100) on a 100 mm scale. VAS scores for study inclusion were between 40 and 90 mm, and the minimal clinically important difference (MCID) for VAS following an intervention or treatment is commonly reported in the range of –18 to –23 mm.16,17 For this study, we considered the MCID to be at least –20 mm.18
Secondary efficacy evaluation included the Oswestry Disability Index (ODI)19 and the EQ-5D Health Index,20 as well as imaging data (magnetic resonance imaging [MRI] and plain film assessments; disc volume was evaluated by an independent imaging core lab), nonsurgical or surgical interventions after the investigational treatment (time to lumbar spine intervention), test of mobility, assessment of exercise, and functional biomarkers of pain. The MCID for the ODI has reported values in the range of 9 to 13.16,18,21–23 For the purposes of this study, MCID of –15 was selected. The MCID for the EQ-5D was considered to be 0.08.24
MRI-based evaluations, from both automated and manual imaging analysis, including mean T2 values, disc volume, disc height, and/or disc height index, Modified Pfirrmann grade, T1 rho, Modic changes, annular tears/fissures, endplate integrity, and herniation, were performed at screening (baseline) and at 12, 26, 52, 78, and 104 weeks. X-ray assessments for disc height and/or disc height index and Kellgren-Lawrence score of disc degeneration were performed at these same intervals.
Safety
An adverse event (AE) was defined as the development of an undesirable medical condition or the deterioration of a preexisting medical condition following or during exposure to the treatment, whether or not considered causally related to the treatment. Safety assessment included documentation of all AEs, including pain level significantly worsened as measured by an increase of more than 30 points on the VAS and function level significantly worsened as measured by an increase of more than 30 points on the ODI compared with baseline. Safety analysis also included assessment of hematology, blood chemistry, serum panel reactive antibodies, vital signs, physical condition, and body weight/height measurements.
AEs were recorded including intensity (mild, moderate, or severe) and causality (not related, possibly related, or probably related). Serious adverse events (SAEs) were defined by standard FDA clinical trial reporting criteria. AEs were considered for the relationship to the injection device, to the injection technique/procedure, and to the investigational cells and/or vehicle. All AEs were classified using MedDRA (Medical Dictionary for Regulatory Activities).
Procedures
Up to 30 days prior to treatment, patients consented and were screened for study inclusion, including baseline MRI and x-ray imaging, for which a central image analysis vendor provided confirmation of study eligibility prior to randomization. Patients eligible to enter the study were randomized via a web-based software platform (Interactive Web Response System) to 1 of 4 treatment cohorts. Randomization occurred between 7 and 10 days before the target treatment date. The study used a double-blinded protocol, with an injecting investigator and an assessing investigator. The injecting investigator could not be blinded due to differences in viscosity of the injected materials, but that investigator did not participate in clinical follow-up. The assessing investigator was blinded to treatment and was not present in the room during investigational product preparation or administration. All patient assessments were conducted by the noninjecting/assessing investigator. Patients and all staff involved in the care of the patients were blinded to treatment. There were 2 parts to the study. In Part 1, patients were randomized in a 4:1:1 ratio to low-dose IDCT (3,000,000 cells/mL; N = 20), vehicle (N = 5), or placebo (saline, N = 5) groups, respectively. Part 2 started after the completion of the enrollment of Part 1. In Part 2, additional patients were randomized in a 4:1:1 ratio to high-dose IDCT (9,000,000 cells/mL; N = 20), vehicle (N = 5), or placebo (saline, N = 5), respectively.
To monitor the safety of the investigational product, the first 6 patients of Part 1 (low-dose, vehicle, and placebo) were enrolled with a separation of 72 hours between dosing. Since no safety concerns were noted, the remaining Part 1 patients were enrolled. Only after Part 1 patients had been fully enrolled, with no safety issues observed and after a separation of 4 weeks, did enrollment for Part 2 (high-dose, vehicle, and placebo) begin, also with a separation of 72 hours for the first 6 patients. To maintain the blinded nature of the study, the control cohorts were enrolled alongside the low- and high-dose cohorts. An independent data monitoring committee monitored the risk-benefit ratio and could recommend stopping dosing.
On day 1 of the study, patients received a single intradiscal injection of 1 mL of their assigned treatment into a single symptomatic lumbar (L3–S1) intervertebral disc under fluoroscopic (or computed tomographic) guidance and local anesthesia or conscious sedation per site standard of care. They remained at the site until meeting discharge criteria. On days 2, 3, or 4 of the study, patients were contacted by phone to determine their condition and whether there were any issues or AEs. At site visits (weeks 4, 12, 26, 52, 78, and 104), patients were screened for standard laboratory parameters, vital signs, pain medication use, disc interventions unrelated to the index injection in the study, VAS for low back pain, ODI, and EQ-5D questionnaires. Timed Up and Go test, assessment of exercise, MRI, radiographs, and neurological examination were also performed to evaluate for primary, secondary, and exploratory endpoints. Serum panel reactive antibodies were drawn prior to administration of treatment and at week 4.
All patients were intended to be followed for a total of 2 years, with a primary study period (1 year) followed by an additional 1-year extension period. After all patients had completed the primary study period at 1 year, the study was unblinded to conduct the primary safety and efficacy analyses. The sponsor, contract research organization (with the exception of blinded site monitors), and Medical Monitor were unblinded at this time. Patients, site personnel (including assessing investigators), imaging vendor, and blinded site monitors remained blinded to individual cohort assignments until after completion of the extension period. The independent data monitoring committee oversaw the clinical study. Except for patients who withdrew from the study, all were followed until study end (104 weeks).
Data Analysis
Interim (week 52) and final (week 104) data analyses were performed. The primary efficacy endpoint was a change from baseline in low back pain at 52 weeks as measured on a 100 mm VAS. A right-tailed, paired t test at a 5% significance level was performed on percentage change from baseline in VAS score at 52 weeks for each treatment group, with the null hypothesis that mean percentage reduction from baseline was less than or equal to 30% vs the alternative that mean percent reduction from baseline was greater than 30%.
Only patients with complete data were included in the primary analyses. To avoid potential biases due to differential drop-out rates, a repeated-measures mixed-effects linear model was used, which uses maximum likelihood to account for the missing data. Change from baseline in VAS score was considered as the response variable, the patient was considered as a random explanatory variable, and visit and a treatment-by-visit interaction were considered as fixed variables. Differences between groups were assessed with a Wald test of treatment.
In addition, the proportion of patients achieving >30% reduction in VAS at week 52 was determined along with a 2-sided exact (Clopper-Pearson) 95% CI. The null hypothesis of no between-treatment differences was tested using Fisher’s exact method.
For secondary efficacy, the changes from baseline in ODI and EQ-5D scores at each time interval were evaluated using 2-tailed paired t tests for each treatment group separately. Analysis of covariance was used to test for differences between groups. For quantitative MRI assessment parameters, baseline values were compared against the values obtained at each time point posttreatment for each treatment group using a 2-sided paired t test.
Results
Primary Efficacy
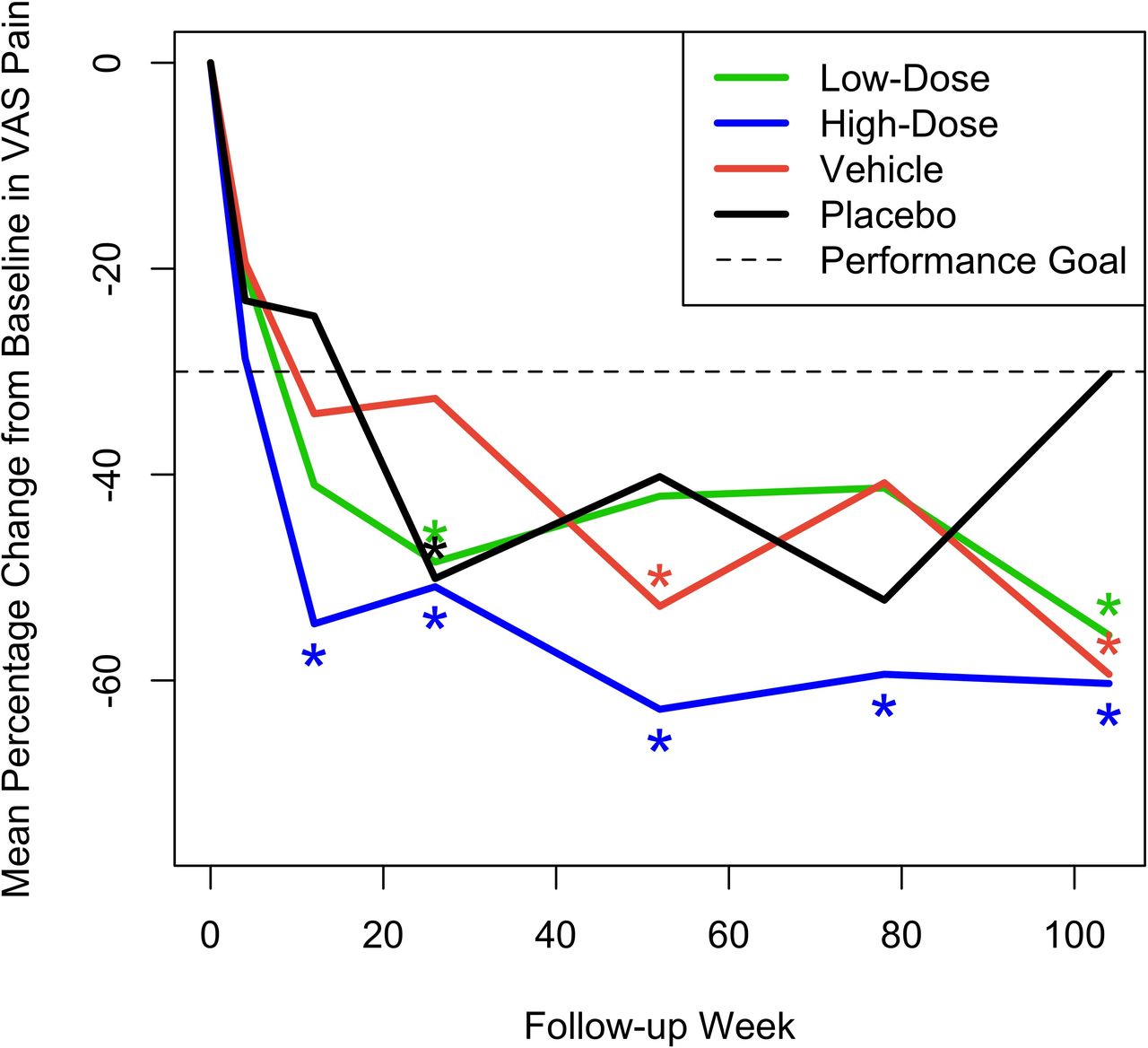
Mean pain VAS measurements on day 1 prior to treatment were as expected for a study population with CLBP (range 60.5–64.9 mm), with no notable difference between treatment groups. At week 52, the study’s primary efficacy endpoint, a statistically significant mean percentage decrease from baseline in VAS, was observed in the high-dose IDCT group (–62.8% [90% CI –77.1, –48.5], P = 0.0005), achieving the primary efficacy endpoint of back pain improvement greater than 30% at 52 weeks. A smaller, but significant percentage decrease in VAS was observed in the vehicle group (–52.8% [90% CI –74.73, –30.92], P = 0.044). VAS scores in the high-dose cell group were also significantly improved over baseline, with the percentage decrease of >30%, at all follow-up intervals after 12 weeks. The vehicle group improved significantly (>30%) in VAS only at week 52 and the low-dose and placebo groups only at week 26 (–48.49% [90% CI –61.03, –35.94], P = 0.010 and –50.13% [–65.24, –35.02], P = 0.021, respectively). Neither of them improved significantly at week 52. Only the high-dose cell group showed a reduction in back pain VAS that was significantly greater than an MCID of –20 mm at week 52 (–42.8 [–55.9, –29.7], P = 0.002) and maintained this significance at all posttreatment intervals. The mean percentage change across time for the 4 groups is shown in Figure 2. Results from the mixed-effects models were similar. In addition, Table 3 provides actual mean VAS values for each treatment group at all test intervals. There were neither statistically significant differences in mean VAS change from baseline across treatment groups at any interval nor were the percentages of patients with >30% reduction in low back pain VAS at week 52 significantly different across groups, although the high-dose IDCT group had the highest percentage of patients with >30% reduction (70%) compared with 55% in the low-dose group and 60% in the combined vehicle and placebo groups.

Mean percent change across time from baseline in visual analog scale (VAS) pain scores for the 4 treatment groups. Asterisks denote statistically significant improvement of greater than 30%.
Mean (SD) pain visual analog scale scores in mm for the 4 treatment groups across time.
Secondary Efficacy Measures
Patient Questionnaires
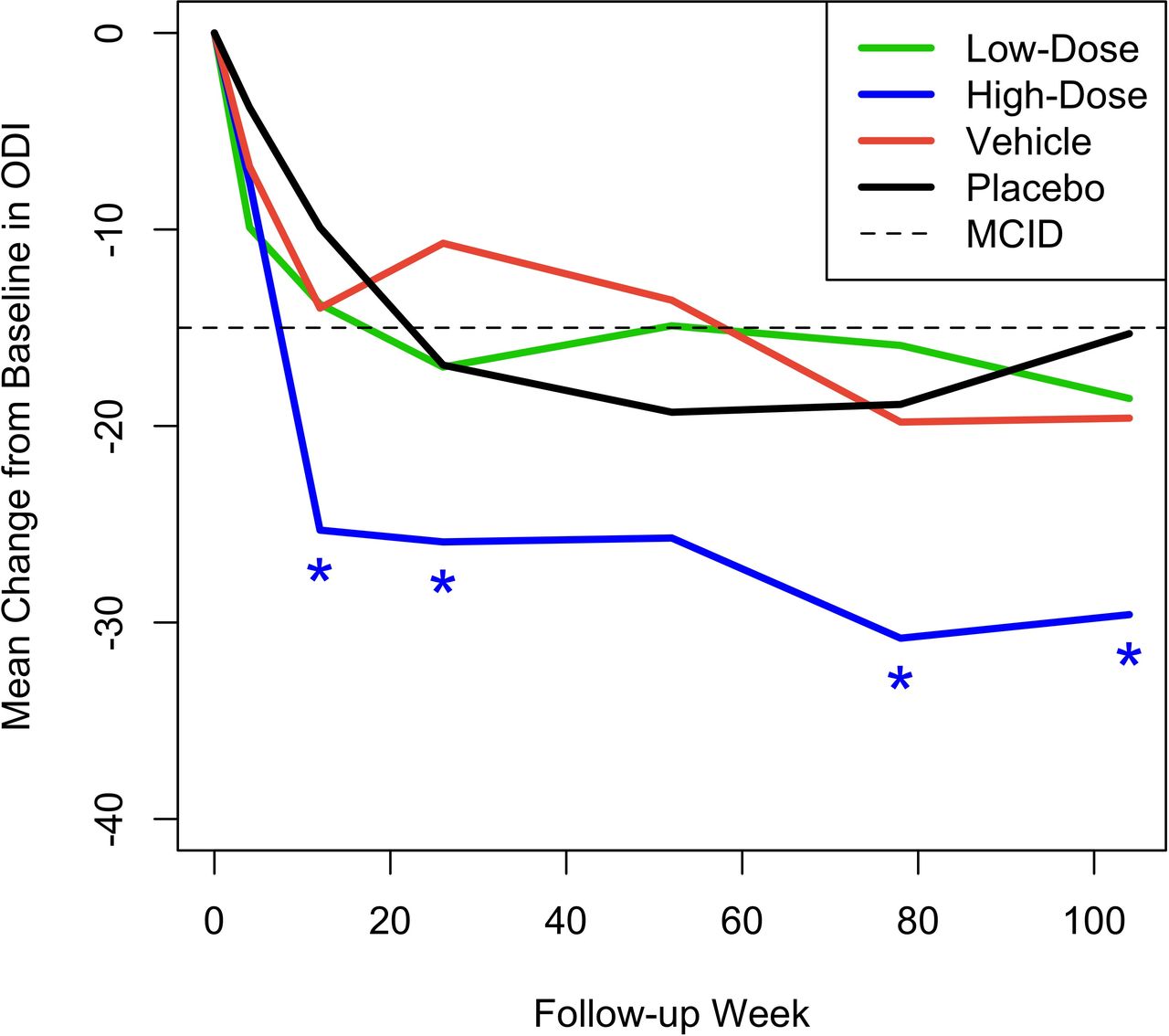
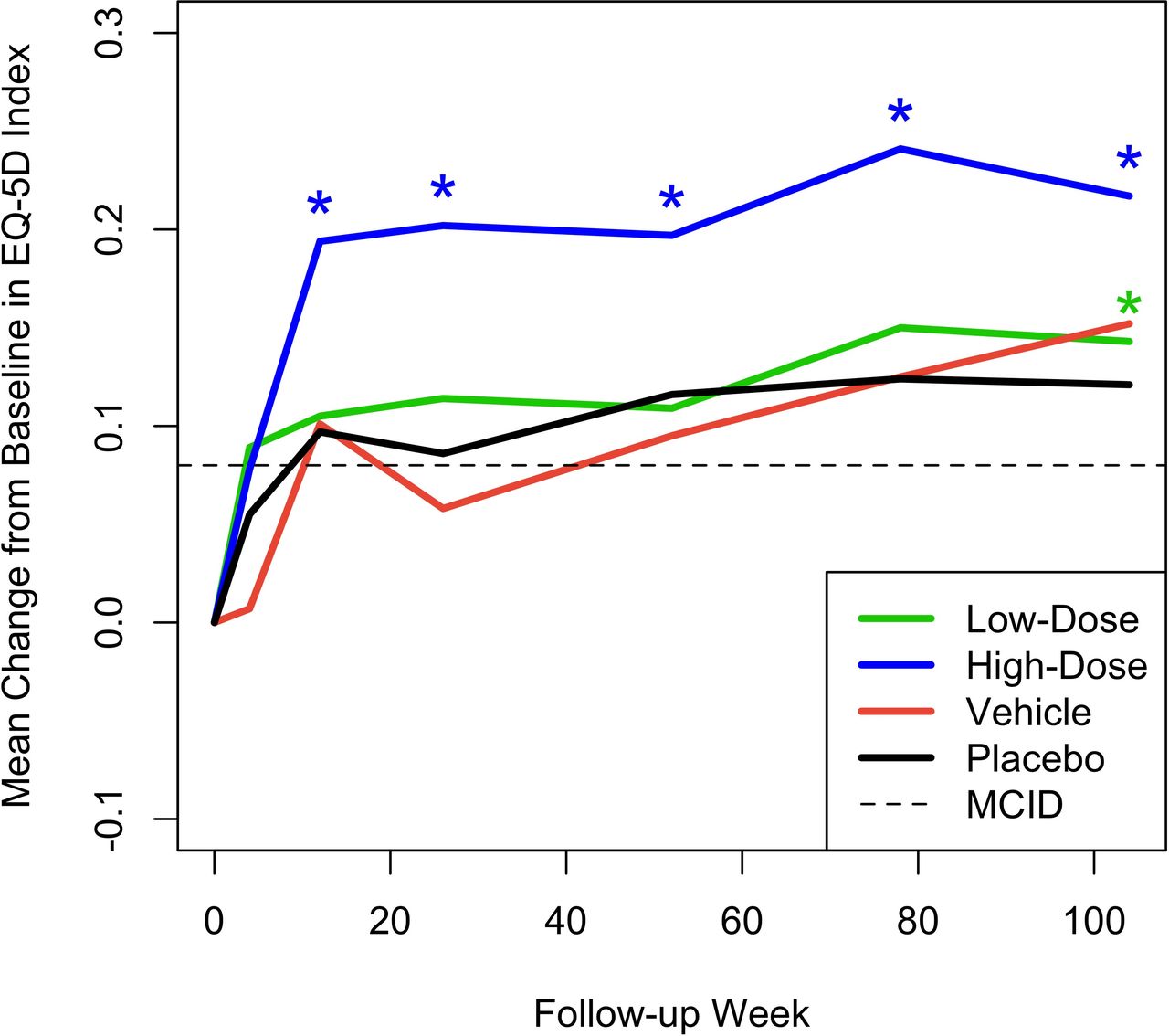
Figures 3 and 4 show the mean change from baseline over time in ODI and EQ-5D, respectively, compared with MCID. Although statistically significant mean improvements from baseline in ODI were observed for all 4 treatment groups, only the high-dose IDCT group demonstrated a mean change that was statistically greater than the MCID at weeks 12, 26, 78, and 104. Importantly, the high-dose IDCT group decreased from the “severe disability” range to the “minimal disability” range by week 78; this improvement was maintained at week 104. The mean change from baseline in EQ-5D index score was statistically significantly greater than the MCID of 0.08 in the high-dose IDCT group, beginning at week 12 (0.194 [0.116, 0.272], P = 0.008) and maintained to week 104. The low-dose IDCT group exhibited improvement in EQ-5D score greater than the MCID at week 104 (0.143 mm, P = 0.023).

Mean change from baseline across time in Oswestry Disability Index (ODI) for the 4 treatment groups. Asterisks denote statistically significant improvement compared with a minimal clinically important difference (MCID) of –15.

Mean change from baseline across time in EQ-5D Health Index for the 4 treatment groups. Asterisks denote statistically significant improvement compared with a minimal clinically important difference (MCID) of +0.08.
Radiology
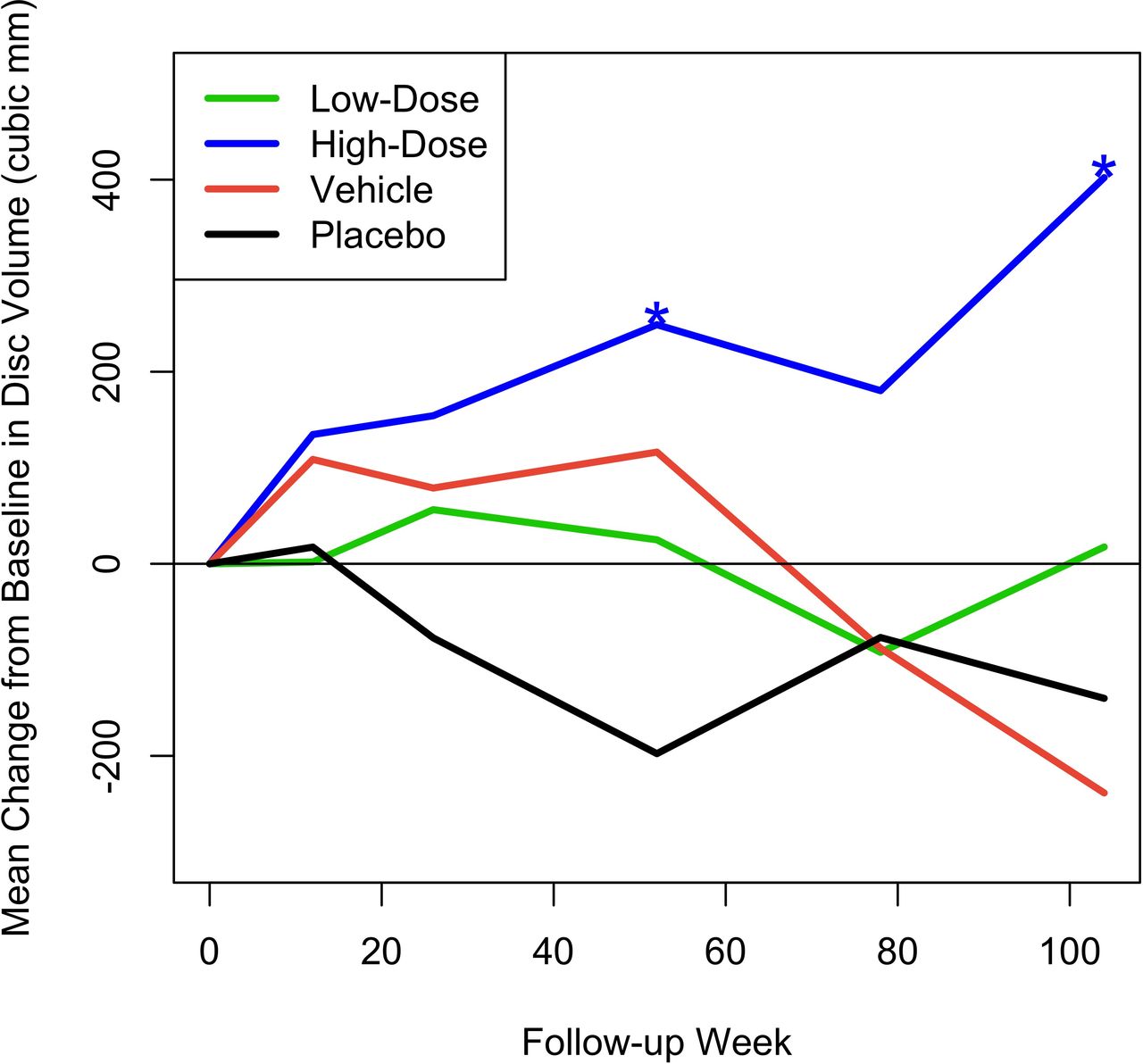
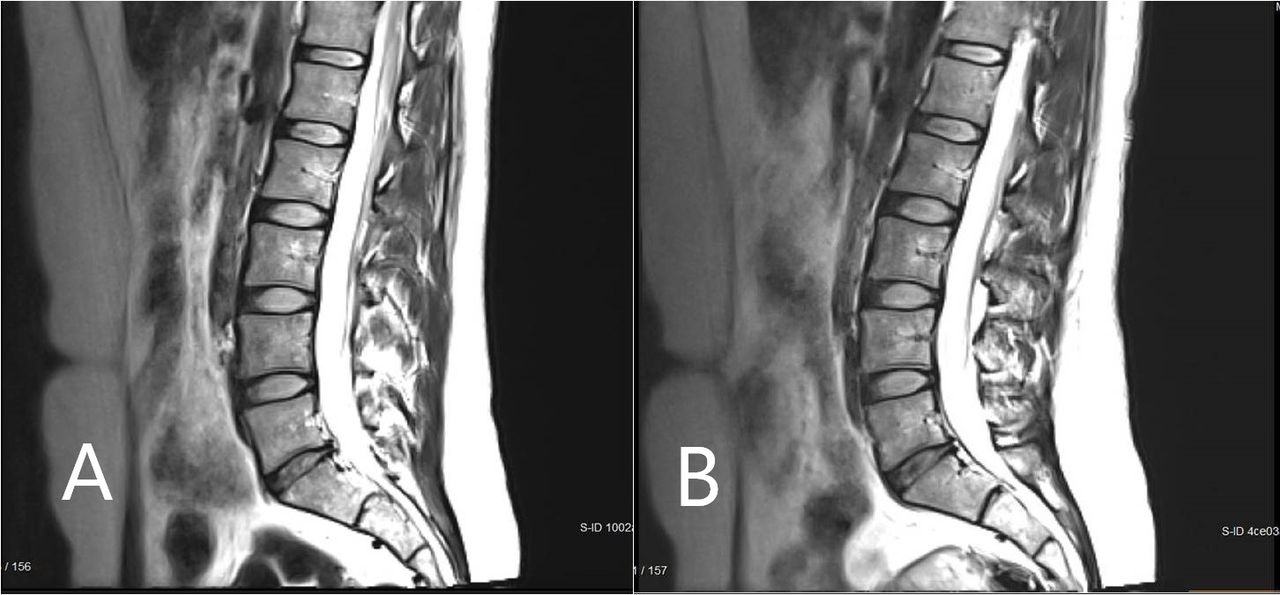
Figure 5 shows the mean change from baseline in MRI measurement of disc volume. This was calculated by an independent imaging core laboratory using a validated, automated method. Only the high-dose group had a statistically significant change over baseline, with volume increase reaching significance at the 52-week endpoint (mean change = 249.01 mm3, P = 0.028) and continuing to increase further by the 104-week final assessment (mean change = 402.12 mm3, P = 0.028). Figure 6 presents an MRI scan of a 35-year-old woman with single-level disc pathology at L5-S1, a posterior annular tear, and loss of disc height seen 14 years after a motor vehicle accident. Disc height improved at 6 months after high-dose IDCT and disc volume clearly increased.

Mean change from baseline in disc volume (mm3). Asterisks denote statistically significant change.

Magnetic resonance imaging scan of a study participant with single-level disc pathology at L5-S1 at (A) baseline and (B) 6 months after high-dose injectable disc cell therapy. Disc height and volume increased by 6 months.
Safety
The overall incidence of treatment-emergent AEs reported during the study was lowest in the high-dose IDCT group, highest in the vehicle and placebo groups, and included back pain, injection site pain, hypoesthesia, paresthesia, extremity pain, muscle spasms, and others. The majority of patients with AEs experienced events of mild or moderate intensity. Overall, 26.7% of patients experienced back pain, which was considered to be not related to the study drug (cell therapy), while 25% experienced back pain reported as related to the study drug. VAS and ODI scores did not worsen by >30 points in any study group. A minority (18.3%) of AEs were reported as severe, and a minority of these (6.7% overall) were treatment-emergent SAEs, all of which were experienced by patients in the vehicle (n = 1) or placebo groups (n = 3). There were no deaths during the study and no events that led to study withdrawal. Overall, clinical laboratory values and vital signs did not change notably over time and were similar across treatment groups.
Discussion
Disc degeneration is a chronic, progressive disease that is characterized by the breakdown of extracellular matrix molecules in the disc. This breakdown is due to the depleted capacity of local cells to produce new matrix as well as an imbalance in the anabolic and catabolic influences on disc tissue.7 These changes result in structural disc alterations that may result in acute and chronic pain. IDCT is an allogeneic cell therapy/device combination product that is a mixture of progenitor cells and a viscous delivery vehicle. The active ingredient of IDCT is a live cell population known as discogenic cells, which are human progenitor cells generated from adult intervertebral disc tissue. In this study, treatment with IDCT was effective and well tolerated.
Overall, the most sustained, clinically meaningful reductions in low back pain were observed in the high-dose IDCT group, and this reduction in pain was accompanied by improvements in patient function (indicated by decreased levels of disability), quality of life, and disc volume. In particular, the high-dose IDCT group, and only this group, showed a reduction from baseline in back pain VAS that was statistically significantly greater than an MCID of –20 mm at week 52. The greatest and most rapidly evolving changes from baseline in ODI were observed for the high-dose IDCT group, and these changes were maintained to week 104. Change in ODI was significantly greater than the MCID of –15 points for only the high-dose IDCT group and decreased from the “severe disability” range to the “minimal disability” range by week 78, where it was maintained at week 104. Change in EQ-5D index score was significantly greater than MCID in only the high-dose IDCT group, beginning at week 12 and maintained to week 104.
Statistically significant improvements from baseline in disc volume were only observed in the high-dose IDCT group. Mean disc volume in this group increased steadily from baseline, and this change reached statistical significance at week 52 and week 104. There were no statistically significant differences between treatments in the number of and time to lumbar spine surgical interventions.
Low-dose IDCT also had a therapeutic effect meeting statistical significance for VAS at weeks 26 and 104 and EQ-5D at week 104, although these effects did not occur as quickly or as robustly as was seen with high-dose IDCT. Overall, both the low and high doses of IDCT were safe and well-tolerated, demonstrating that IDCT can be safely injected into a degenerated lumbar disc for the treatment of symptomatic DDD. The few SAEs all occurred in the control groups.
New matrix generation is one of the mechanisms of action for IDCT. The generation of extracellular matrix has been observed in animal studies through histology13 and confocal imaging of cells, as well as via quantitative evaluation of gene expression through real-time polymerase chain reaction. Also, the ability of the cells to generate proteoglycans, which help drive disc hydration, has been quantified using a biochemical assay. This includes the important proteoglycan, aggrecan, which has been measured in a protein assay.15 Discogenic cells are allogeneic, as they are derived from adult human organ donors. However, we have observed that discogenic cells appear to be immune-privileged. First, human discogenic cells have been implanted into animals, and no immune response has been noted despite a lack of immune suppression in these animals. Second, discogenic cells have been combined with peripheral blood mononuclear cells in mixed lymphocyte reactions, and minimal concomitant peripheral blood mononuclear cell proliferation was noted (which is consistent with a lack of rejection/immune response). Two possible mechanisms for this immunological evasion are proposed. First, the cells are major histocompatibility complex class I positive (HLA-ABC) and major histocompatibility complex class II negative (HLA-DR/DP/DQ), making them hypoimmunogenic. Second, discogenic cells lack expression of co-stimulatory molecules CD40, CD80, and CD86, which are required for effector T cell induction.13,15 The cells do not form tumors in vitro or in vivo. Studies testing human IDCT in animal models of disc degeneration have shown IDCT to be safe and bioactive.
The primary limitation of this study was the relatively small number of patients, as it was a Phase I/II industry-sponsored clinical trial. Even so, statistically significant differences were consistently observed over a variety of outcome measures. A Phase III clinical trial is scheduled to begin in 2024.
Conclusions
In this combined Phase I and Phase II FDA-approved clinical trial, high-dose allogeneic disc progenitor cell therapy was safe, was well tolerated, and significantly increased disc volume. The high-dose patients had clinically meaningful, statistically significant improvement beyond MCID in VAS, ODI, and EQ-5D by 12 weeks. Clinical improvement was sustained at 6 months, 1 year, 1.5 years, and 2 years following a single intradiscal injection. These data suggest that high-dose IDCT may be a safe and effective treatment for symptomatic DDD, producing the most rapid, sustained, and clinically meaningful outcomes in this study. In the low-dose IDCT group, there was a trend in the improvement of clinical outcomes, though this finding was inconsistent. While the vehicle resulted in some pain relief, it was not associated with clinically meaningful improvements in function or quality of life. No consistent or durable statistically significant or clinically meaningful outcomes were observed in the placebo group. The promising results of high-dose IDCT in this study, particularly the sustained decrease in low back pain and improvements in function, quality of life, and disc volume, support further development of this therapy.
Supplementary material
online supplementary file 1.
Acknowledgments
Karen I. Berliner, PhD, helped with manuscript preparation and editing.
Footnotes
Funding The clinical trial was sponsored and funded by DiscGenics, Inc., Salt Lake City, UT.
Declaration of Conflicting Interests Dr. Foley discloses that he has a patent relationship to the product in this study, stock options, and is the Chief Medical Officer and on the Board of Directors of the study sponsor. Drs. Beall, Davis, and DePalma report research funding from the sponsor as investigational sites. Dr. Kelly was the independent statistical consultant to the study sponsor for this study. None of the other authors report conflicts of interest related to this study. Additional unrelated financial disclosures and conflicts of interest are listed in the supplementary file 1.
IRB Approval By sponsor—Advarra IRB, assurance #00023875. Each investigator site also obtained IRB approval.
- This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery. Copyright © 2024 ISASS. To see more or order reprints or permissions, see http://ijssurgery.com.
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.