


Abstract
While achieving premarket approval from the US Food and Drug Administration represents a significant milestone in the development and commercialization of a Class III medical device, the aftermath endeavor of gaining market access can be daunting. This article provides a case study of the Barricaid annular closure device (Barricaid), a reherniation reduction device, which has been demonstrated to decrease the risk of suffering a recurrent lumbar intervertebral disc herniation. Following Food and Drug Administration approval, clinical adoption has been slow due to barriers to market access, including the perception of low-quality clinical evidence, questionable significance of the medical necessity of the procedure, and imaging evidence of increased likelihood of vertebral endplate changes. The aim of this article is to provide appropriate examination, rationale, and rebuttal of these concerns. Weighing the compendium of evidence, we offer a definition of a separate and unique current procedural terminology code to delineate this procedure. Adoption of this code will help to streamline the processing of claims and support the conduct of research, the evaluation of health care utilization, and the development of appropriate medical guidelines.
Introduction
Bringing a new medical device technology to market in the United States (US) can be a protractive and often arduous process.1 This is particularly true for novel devices that do not have a predicate and introduce a unique indication for use. Consequently, these technologies are rare and subject to the most stringent level of regulatory oversight (Class III) by the Food and Drug Administration (FDA) and require premarket approval (PMA).2
Even with extensive experience, organization, and attentiveness, most medical device companies are ill-prepared for the exigencies they will encounter in eventually gaining market access and widespread clinical adoption.3 So much effort and time are expended in fulfilling the preclinical and clinical requirements necessary to achieve PMA, that limited energy, financial resources, and, we dare say, patience are left to tackle the daunting task of actually introducing the device into the marketplace for physicians to utilize and patients to benefit from. Indeed, the costs associated with the entire research, development, and commercialization processes of bringing a new technology to market have been estimated in excess of US$500 million.4
In the US health care system, device manufacturers, post-PMA, are faced with the complexities of the reimbursement universe, which comprises an assortment of stakeholders and decision-makers who include professional medical societies, private insurance providers, the Centers for Medicare and Medicaid Services, the American Medical Association, and others. The gauntlet to obtaining an American Medical Association Current Procedural Terminology (CPT) code specific to a novel indication for use, and subsequently undergoing a determination of the resources required to provide an explicit medical service via the Relative Value Scale Update Committee, can be fraught with setbacks and requires perseverance and political savvy.
This article provides a case study of the Barricaid annular closure Device (ACD; Barricaid), a reherniation reduction device, which has been demonstrated to decrease the risk of suffering a recurrent lumbar intervertebral disc herniation. Herein, we discuss the barriers to market access encountered with the introduction of this spinal device and provide the rationale for clinical acceptance and adoption.
Recurrent Lumbar Disc Herniation
Perhaps the earliest clinically significant manifestation of degeneration of the lumbar spine is loss of structural competence of the annulus fibrosus of the intervertebral disc, which increases the risk of extrusion of central nucleus pulposus.5 The resultant herniation exposes the disc material to noxious stimuli, promotes an inflammatory reaction of the adjacent nerve roots, and directly compresses the lumbosacral nerves by extruded disc material.6 This multifactorial mechanism of pain generation involves components of low back pain coupled with the cardinal feature of lower limb radiculopathy.7 When symptoms are chronically severe and resistant to conservative measures, surgical discectomy remains a viable treatment option.8 In fact, surgery for lumbar disc herniation is the most common indication for performing spinal surgery.9
While discectomy can effectively ameliorate symptoms associated with disc herniation, the resultant surgical defect substantially diminishes the structural integrity of the annulus, placing the disc at risk for reherniation. This postdiscectomy complication is not uncommon, and recurrent disc herniation is the primary cause of surgical failure and morbidity in patients treated with lumbar discectomy.10–13 Patients having a recurrent disc herniation can experience re-emergence of pain and functional deficits of greater severity than the index herniation, are less likely to return to work, and spend more days in the hospital than patients without reherniation.13 Diagnosis and management of this subset of patients also place a significant burden on the health care system with aggregate costs that are many fold higher than in unaffected patients.14
Revision surgery to correct the reherniation is decidedly more complex and less successful than the primary discectomy procedure.13 Altered anatomy from the previous surgery and epidural scarring create an unfavorable surgical environment and increase the risk of dural tears and hemorrhage. A wide dissection and extensive bone removal with aggressive facetectomy are often required for visualization and to provide satisfactory decompression of the neural foramen. If extensive decompression is involved, then instrumented spinal fusion may be warranted to provide necessary stability to the motion segment, adding further costs and resource use to the treatment plan.11 It has been estimated that 53% of reoperations involve a fusion procedure.15
A major determinant of recurrent disc herniation is the size of the residual defect following the initial discectomy procedure,16,17 with defects ≥6 mm in width showing the greatest susceptibility to reherniation.18 Recognizing the clinical importance of preventing recurrent disc herniation and the associated revision surgery, the Barricaid device was developed to specifically address patients with large annular defects at the highest risk for reherniation and poor outcomes, reoperations, and often multiple repeat surgeries.
Barricaid: Technical Description
The Barricaid serves as an adjunct to the discectomy procedure and is surgically implanted into the residual annular defect at the conclusion of the operation. The implant is permanent and has 2 major subcomponents: a flexible woven polymer fabric component that is intended to occlude the annular defect, and a titanium bone anchor that affixes the flexible polymer component in place (Figure 1). The titanium component is anchored to healthy bone of an adjacent vertebral body. Barricaid is designed to withstand 330 psi of pressure in the spinal disc, roughly 10 times the pressure in a standard car tire.19 It also allows for normal kinematics and physiological movements of the affected spinal motion segment following surgery.

The Barricaid annular closure device.
The Barricaid system is available in 2 implant widths, 8 and 10 mm, to accommodate variations in annular defect size. The implant is preloaded onto disposable delivery tools.
Surgical Technique—Limited Discectomy
A key feature of the Barricaid implantation procedure is that it allows for less aggressive discectomy and maximum preservation of disc material. Conservation of disc height and integrity is integral to averting spinal degeneration at the affected motion segment, particularly facet arthropathy. The limited discectomy procedure is summarized briefly as follows:
Upon achieving sufficient exposure of the lateral wall, the dural sac and nerve root are carefully retracted toward the medial side to access the affected disc area. Any free fragments are extracted without making an incision into the annulus. For extruded disc herniations, the protruding part is meticulously excised, and any adjacent loose fragments are cleared using a small pituitary rongeur. In situations where the disc is merely protruding, an incision is made in the annulus followed by the thorough removal of all loosened disc material. The procedure concludes with a careful evaluation of the neural foramen after disc removal followed by the insertion of the implant (Figure 1).
Clinical Effectiveness
The safety and effectiveness of the Barricaid device were demonstrated in a multicenter, randomized pivotal trial conducted between December 2010 and October 2014 (NCT01283438). A total of 554 patients with large annular defects (6–10 mm) were randomly assigned to receive treatment with discectomy and annular closure or discectomy alone.20 At the 2-year primary study follow-up, the a priori–defined study endpoints were met favoring the ACD.21 The frequency of symptomatic reherniation was significantly lower with the ACD (12% vs 25%, P < 0.001). Likewise, the frequency of reoperations to address recurrent disc herniation correspondingly favored patients treated with annular closure (5% vs 13%, P = 0.001). These findings have been confirmed in subsequently conducted randomized controlled trials.22,23
There have been 3 recently published meta-analyses that have been performed to determine the global effectiveness of ACDs for preventing the occurrence of intervertebral disc reherniations and associated reoperations.24–26 The pooled data analysis showed that the addition of annular repair at the conclusion of the discectomy procedure significantly reduced the rate of recurrence and reoperation compared with lumbar discectomy alone. Specifically, the use of the Barricaid device was found to be most effective in preventing reprotrusion and reducing reoperation rates.26
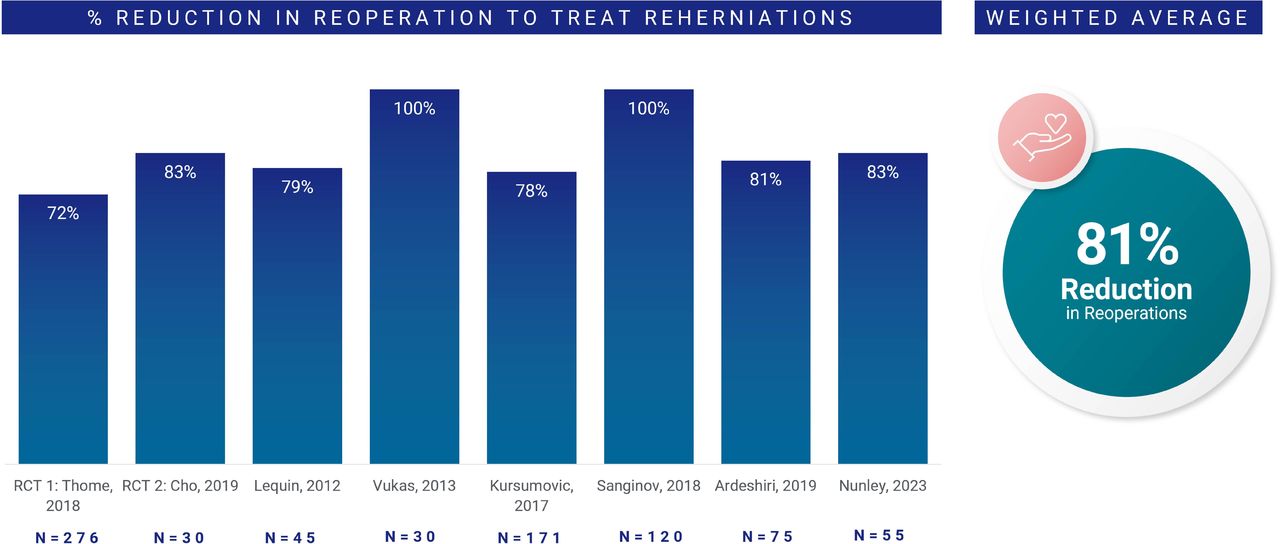
Figure 2 illustrates the weighted average of percentage reduction in reoperations for recurrent disc herniation in approximately 800 patients drawn from 8 distinct study populations.21–23,27–31 There was a weighted 81% average reduction in reoperations across these groups (range: 72%–100%).
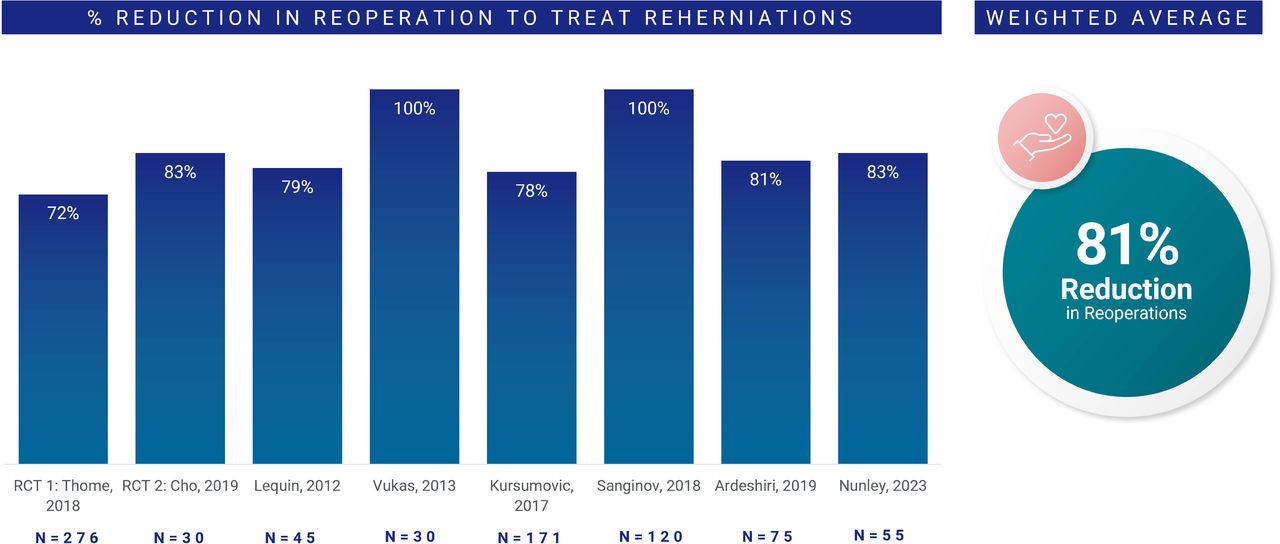
Weighted average of percentage reduction in reoperations for recurrent disc herniation across 8 distinct studies (~800 patients). Values at a minimum postoperative follow-up of 1 year.
Regulatory Status
The date of the FDA Notice of Approval for the Barricaid device was 8 February 2019 (P160050). Thus, Barricaid is the only ACD to receive PMA from FDA specifically indicated for reducing reherniation and reoperation after lumbar discectomy and is the only ACD approved by the FDA for use in the spine. Barricaid remains the only ACD on the US market.
The Barricaid ACD is indicated for reducing the incidence of reherniation and reoperation in skeletally mature patients with radiculopathy (with or without back pain) attributed to a posterior or posterolateral herniation and confirmed by history, physical examination, and imaging studies, which have demonstrated neural compression using magnetic resonance imaging to treat a large annular defect (between 4 and 6 mm tall and between 6 and 10 mm wide) following a primary discectomy procedure (excision of herniated intervertebral disc) at a single-level between L4 and S1.
Barriers to Clinical Adoption
The FDA-approved Barricaid device, with its unique indication for use, has faced significant barriers to clinical adoption in the US. The device has been evaluated by 2 separate technology assessments (Hayes, ECRI) as a means of guiding stakeholders in their analyses and assessments of clinical utility. These reports have concluded that the quality of evidence to support reimbursement is low, and there has been only one national coverage decision that covers Barricaid as an adjunctive treatment for recurrent disc herniation.
Reimbursement Status
Current reimbursement for the Barricaid procedure in the US is mostly determined on a case-by-case basis with prior authorization. Cigna initiated coverage of the Barricaid device procedure as of 15 June 2023.
Device Classification
Point
Technology assessments of annular closure have described the Barricaid device as experimental or investigational.
Counterpoint
A new Class III medical device is considered investigational while it is being evaluated by the FDA for safety and effectiveness under the aegis of a pivotal trial to support PMA.32 These data, along with supporting preclinical findings, are then presented to the FDA in the form of a PMA application. In 2019, Barricaid received approval by the FDA for the indications for use specified in the Regulatory Status section. Thus, this indication for use is no longer considered investigational for the FDA-approved Barricaid device.33 To ensure that the Barricaid device continues to perform according to the approved indication, the FDA has mandated a standard 10-year postmarket assessment period to monitor device safety and effectiveness in real-world clinical settings. To date, the device has been implanted in more than 1500 patients in the US with corroborative evidence of device safety and effectiveness. While the device may not yet be a well-established adjunctive procedure with discectomy, its safety and effectiveness for the FDA-approved indication for use have been proven.
Medical Necessity
Point
The defined medical problem—reherniation following primary discectomy—does not constitute a serious clinical sequela.
Counterpoint
Recurrent disc herniation qualifies as a serious postoperative complication based on the FDA’s adverse event classification system, which includes, in part, inpatient hospitalization or prolongation of existing hospitalization and a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions. This adverse event occurs commonly following primary discectomy with estimates ranging from 5% to 18%.10 In the Barricaid pivotal trial, 25% of discectomy-treated patients suffered a symptomatic reherniation.21 Additionally, revision surgery necessary to correct symptomatic reherniations is a difficult procedure even for a skilled spine surgeon and may require conversion to instrumented spinal fusion if extensive decompression is involved.
Device Integrity
Point
The Barricaid device is associated with postoperative device-related adverse events, which may include device breakage, malfunction, and/or dislodgment.
Counterpoint
An evaluation of the pivotal trial patient population through 5 years of follow-up found that serious adverse events related to the procedure or device were less frequent for Barricaid than discectomy alone (P = 0.008). Additionally, the incidence of reoperation for any reason (including device issues) was significantly lower with Barricaid compared with discectomy alone (P = 0.03).34
Methodology Concerns
Point
The Barricaid data generated from the pivotal trial and other studies are of low methodological quality due to the lack of blinding, as well as author bias, inclusion of only large defects, bias in reoperation rates, and a 75% long-term follow-up rate. Additional confirmatory randomized trials (with blinding) are warranted.
Counterpoint
The FDA required a single pivotal randomized controlled trial to allow for their determination of safety and effectiveness for the prescribed indication for use, which is restricted to large annular defects (6–10 mm width). The primary trial endpoints were met favoring the ACD, and the FDA did not request that additional clinical studies be conducted to augment their determination of safety and effectiveness or their decision to grant approval.
Most grading systems of the methodological quality of randomized controlled trials are based on the pharmaceutical model of clinical research where blinding is weighted heavily in the quality score.35 However, in drug trials with the use of placebo medications, blinding of patients, investigators, and evaluators is fairly simple.36 Medical device surgical trials, in sharp contrast, are almost impossible to blind adequately, and very few sham-controlled trials of orthopedic surgical procedures have ever been successfully conducted. Consequently, these studies invariably score low on standard assessments of methodological quality.
While blinding remains an important methodological characteristic of study design to protect against bias, its impact is most strongly associated with subjective patient-reported outcomes such as the perception of pain and its intensity where the placebo effect can be demonstrative.37–39 Knowledge of treatment group assignment, on the other hand, likely has very little association with more objective outcomes such as the occurrence of reherniation.40
Thus, dismissing robust clinical evidence and requesting that additional trials be conducted with the false expectation that they can be blinded are a red herring. Despite these inherent blinding challenges, a subset of patients in the Barricaid pivotal trial were blinded to their treatment, and analyses demonstrated that the positive clinical outcomes associated were the same, regardless of blinding.41
Morphological Sequelae
Point
The frequency of endplate changes associated with the Barricaid device is significantly higher than it is with discectomy alone and represents a serious adverse event.42
Counterpoint
Vertebral endplate changes observed with the ACD are morphologically indistinguishable from intravertebral disc herniations or Schmorl’s nodes as described in the classic article by Resnick and Niwayama.43 These areas of herniation occur commonly in the degenerative lumbar spine with a reported prevalence as high as 76%.44 Weiner et al45 reported a vertebral endplate lesion prevalence of 43% following discectomy without evidence of clinical sequelae. Because these changes are almost uniformly asymptomatic, they are detected only as incidental radiographic findings on magnetic resonance imaging scans being conducted for other medical issues.46 No consensus on pathogenesis exists.47
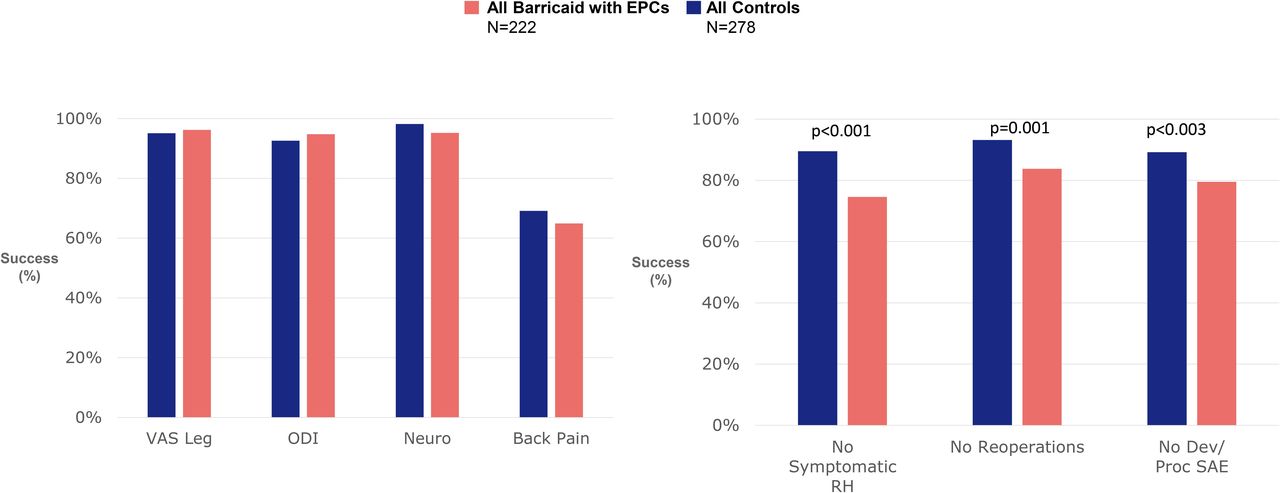
Findings from the pivotal trial confirm the lack of clinical significance of incidental endplate changes. No significant associations were observed between the presence of endplate changes and patient-reported clinical outcomes. There were no reoperations performed for endplate changes, and no serious adverse events were reported that were specifically linked to endplate changes.48 Figure 3 illustrates the comparative responder rates for patient-reported clinical outcomes, symptomatic reherniations, reoperations, and adverse events between Barricaid-treated patients with evidence of endplate changes and patients treated with discectomy only. These findings reflect the overall trial results with no evidence of clinical sequelae associated with endplate changes. Moreover, in the pivotal trial, Kursumovic et al48 reported that Barricaid patients with endplate changes actually performed better than Barricaid patients without endplate changes in the presence of serious adverse events (5% vs 21%), symptomatic reherniations (10% vs 11%), reoperations (4% vs 11%), as well as leg pain (94% vs 92%), back pain (68% vs 60%), and back function (92% vs 92%) improvements.
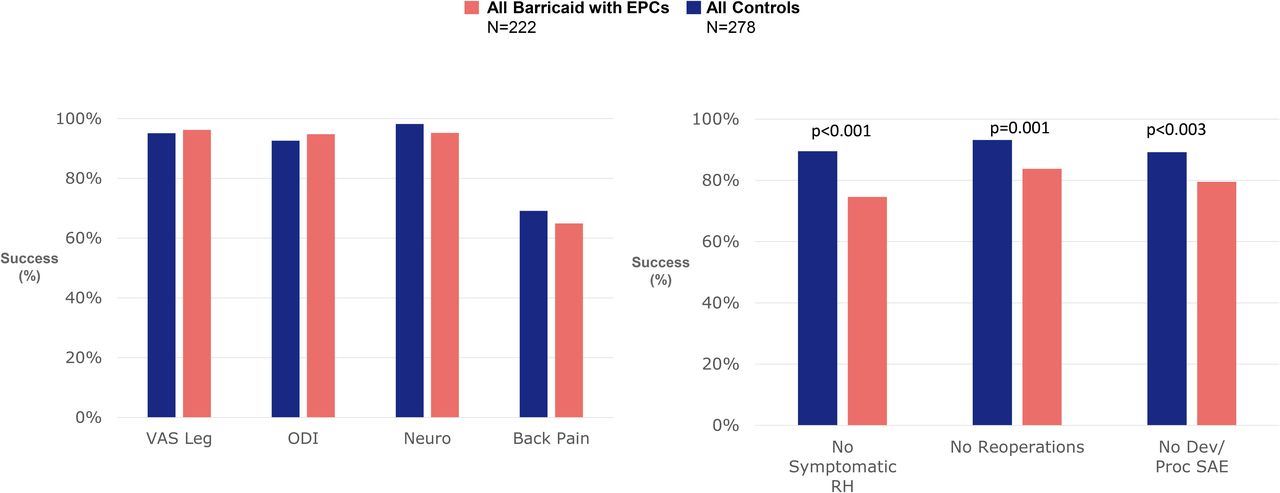
Comparative responder rates for patient-reported clinical outcomes, symptomatic reherniations, reoperations, and adverse events between Barricaid-treated patients with endplate changes and patients treated with discectomy only. Abbreviations: Dev, device; Neuro, neurologic; ODI, Oswestry Disability Index; Proc, procedure; RH, reherniation; SAE, severe adverse event; VAS, visual analog scale.
To explain this seemingly paradoxical finding, the FDA’s current product classification of the reherniation reduction device includes in its description of the technical method that “The device also creates lesion(s) in the vertebral body that may also prevent the herniation of nucleus material out of the annulus.”49
Interpretation
Being the only FDA-approved reherniation reduction device commercially available in the US, the Barricaid has faced significant challenges achieving market access in the medical market, which has hampered clinical adoption. This one-of-a-kind device and procedure are without a comparator, which makes assessments of the clinical importance of the reported reductions in reherniations and reoperations difficult to gauge by stakeholders and decision-makers. Consequently, some payors have been skittish about offering coverage. Having technology assessments judge the underlying data quality as low has further hampered the acceptance of this device as proven technology for its approved indication.
An important feature that differentiates conventional lumbar discectomy from a discectomy procedure augmented with annular closure is the ability to perform less aggressive debulking of the herniated disc material. Sealing the annular defect following a limited discectomy allows for conservation of the maximum amount of nucleus pulposis and has been shown experimentally to restore intradiscal hydrostatic pressures closer to preoperative levels.50 In a randomized controlled trial, Cho et al22 reported significantly better maintenance of disc height in patients treated with annular closure compared with patients having conventional discectomy. These findings may have important downstream clinical implications as preservation of disc height is essential in decelerating the degenerative cascade in the lumbar spine.5,51
As clinical data have accumulated confirming the significant reduction in recurrent disc herniations with annular closure, Cigna Health Care issued a positive coverage policy on 15 June 2023 that enables discectomy patients with large annular defects access to Barricaid. Cigna Health Care represents 1 of the 5 largest commercial payors in the US.
Recommendations
In our opinion, we, the authors of this article, support and recommend the adoption and establishment of unique CPT codes to report laminotomy (hemilaminectomy) with decompression using a new device that requires measurement and alignment of an ACD to treat the removal of disk material between spinal bone components (vertebral bodies).
Specifically, we propose the establishment of a single Category I code to report a new type of decompression procedure that not only utilizes many of the elements of current, “conventional” laminotomy/hemilaminectomy procedures but also includes use of a device to measure and treat the residual disk-removal defect. The procedure includes many of the decompression components noted in existing laminotomy/hemilaminectomy decompressive procedures—freeing of the nerve roots, partial facet removal, opening of the foramen, and excision of herniated disc material. In addition, this procedure repairs the annular defect that remains after disc decompression of herniated/problematic intervertebral disc. This is addressed not only by measurement of the annular defect but also by the use of imaging to align the ACD and by examination and removal of any bony material that may be necessary to ensure the proper angle and alignment of the device prior to its attachment. The procedure includes imaging necessary to accomplish the procedure.
Conclusions
Despite FDA approval for the prevention of symptomatic reherniations and associated reoperations following primary discectomy, the Barricaid device has faced significant challenges gaining market access in the US. Barriers to clinical adoption include the perception of low-quality clinical evidence, questionable significance of the medical necessity of the procedure, and imaging evidence of increased likelihood of endplate changes. The aim of this article has been to provide appropriate examination, rationale, and rebuttal of these concerns. Weighing the compendium of evidence, we recommend the establishment of a separate and unique CPT code to delineate this procedure.
Footnotes
Funding J.E.B. received funding to assist in the preparation of this manuscript from Intrinsic Therapeutics, Inc. (Woburn, MA, USA). This research received no additional external funding.
Declaration of Conflicting Interests The authors report no conflicts of interest in this work.
Disclosures Andrew Metzger reports consulting fees from Intrinsic Therapeutics. Betsy Grunch reports research support from Orthofix; royalties/licenses from Hyhte Holdings and Choice Spine; consulting fees from Brainlab, GT Medical Technologies, Nevro, Zimvie, OptimaSurgical, WebMD, Stryker, Corelink, Playback Health, Alevio, Spineology, and Orthofix; payment or honoraria from the Georgia Society of International Pain Physicians and the North American Neuromodulation Society; payment for expert testimony from various law firms; travel expenses from Georgia Society of Interventional Pain Physicians, the North American Neuromodulation Society, and NASS; leadership or fiduciary role for the Scientific Advisory Board for Spineology; stock from Spineology. William Watters reports consulting fees from Intrinsic and Change Health Care; payment for expert testimony from various entitites; participation in a data-monitoring board for Turning Point Health Care Solutions; and a leadership or fiduciary role for NASS and The Spine Journal. Gunnar Andersson reports consulting fees from Orthofix; participation in a data-monitoring board for Agnovos and Spineart; and a leadership or fiduciary role for as well as stock/stock options from Tetrous Inc. The remaining authors have nothing to disclose.
Author Contributions Conceptualization, M.P.L. and J.E.B.; methodology, J.E.B.; software, J.E.B.; validation, K.-U.L. and J.E.B.; formal analysis, J.E.B.; investigation, M.P.L. and J.E.B.; resources, M.P.L., K.-U.L., and J.E.B.; data curation, M.P.L., K.-U.L., and J.E.B.; writing—original draft preparation, J.E.B.; writing—review and editing, M.P.L., W.C.W., B.H.G., A.K.M., K.-U.L., J.E.B., and G.B.J.A.; visualization, J.E.B.; supervision, M.P.L. and J.E.B.; project administration, M.P.L., K.-U.L., and J.E.B.; funding acquisition, M.P.L. and J.E.B. All authors have read and agreed to the published version of the manuscript.
- This manuscript is generously published free of charge by ISASS, the International Society for the Advancement of Spine Surgery. Copyright © 2024 ISASS. To see more or order reprints or permissions, see http://ijssurgery.com.
References
In this issue





{kind=link}
{kind=link}
{kind=link}